|
|
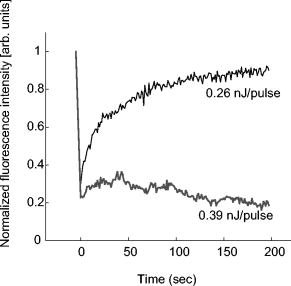
1.IntroductionComplex cell behavior such as cell division, metabolism, and signal transduction is based on a combination of processes at the subcellular level. Imaging and manipulation of intact organelles in a living cell are essential to understanding such complex intracellular dynamics. Fluorescent proteins allow analyses of the dynamics of organelles. When the fluorescent protein is fused with a native protein or a signal peptide, it becomes possible to visualize and track the targeted cellular structures or organelles in real time. The applications of fluorescent proteins continue to provide new insights into various subcellular events in living cells. Focused femtosecond lasers can be used to image intact subcellular structures using multiphoton excitation.1, 2, 3 Femtosecond lasers in the near-infrared region offer attractive advantages, including high spatial resolution, deep penetration into thick samples, and reduced photon-induced damage. Femtosecond lasers are thus suitable for imaging and manipulating intact organelles in living cells. The applications of manipulation of subcellular structures using multiphoton excitation include fluorescence recovery after photobleaching (FRAP),4, 5, 6, 7, 8, 9 intracellular ablation or nanosurgery, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 and photoconversion or photoactivation. 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 In this work, we report on manipulation of intracellular organelles by two-photon excitation. In Sec. 2, we describe a two-photon excitation method for monitoring the dynamics of green fluorescent protein (GFP)-histone in vivo. We investigate the dynamics of histone H1-sGFP in tobacco BY-2 cells by single- and two-photon FRAP. We compare the recovery rate of GFP-tagged histone H1 between single- and two-photon FRAP in tobacco BY-2 cells, and obtain similar results both in HeLa cells and tobacco BY-2 cells. We find the universality of molecular dynamics among species, because the recovery rates are similar both in HeLa and tobacco BY-2 cells. In Sec. 3, we describe the differences between bleaching and intracellular ablation of a mitochondrion using a femtosecond laser oscillator. We show the viability of the cells after femtosecond laser radiation by using propidium iodide (PI). In Sec. 4, we demonstrate marking of mitochondria by using two-photon conversion of the photoconvertible protein, Kaede. Photoactivation and photoconversion enable selective activation and conversion of fluorescence signals from organelles in living cells after focusing femtosecond laser pulses. We demonstrate selective marking of individual mitochondria at different sites in a living BY-2 cell by using two-photon conversion of Kaede, and track of their dynamics. 2.Two-Photon Excitation for Analyzing Molecular Dynamics in Nuclei2.1.Two-Photon Fluorescence Recovery after Photobleaching for Analyzing Histone with High Mobility In VivoThe packaging of eukaryotic DNA is performed by histones, which are mainly classified into H1, H2A, H2B, H3, and H4. The H2A, H2B, H3, and H4 histones are core histones that assemble into an octamer as a nucleosome core particle. The H1 histone is a linker histone that connects one nucleosome core particle to the next. Data regarding the mobility of GFP-tagged histone H1, H2A, H2B, H3 and H4 proteins in living human cells have been reported.40, 41, 42, 43 In those studies, single-photon excitation was used to obtain the mobility. However, photobleaching using single-photon absorption has a disadvantage, namely, collateral absorption outside the focal volume, as mentioned before. Therefore, it is difficult to observe the mobility of a fluorophore along the beam propagation axis ( axis) by single-photon excitation. With two-photon bleaching, the photobleached volume along the axis can be limited.7, 9 We established a HeLa cell line to stably express enhanced GFP (EGFP)-histone H1.2.44 Using this cell line, we measured the dynamics of EGFP-histone H1.2 in a selectively bleached heterochromatic region of the nucleus by single-photon and two-photon FRAP (Fig. 1 ). In the case of single-photon FRAP, the GFP fluorescence of bleached areas recovered within a maximum of , as shown by the representative recovery curve. In contrast, with two-photon FRAP using the same region of interest (ROI) size, the EGFP fluorescence recovered within . Thus, the fluorescence recovery rate for two-photon excitation was much shorter than that for single-photon excitation. The difference in the fluorescence recovery rates was due to differences in the bleaching properties between single- and two-photon excitation. Similar to fixed cells, in living cells, EGFP molecules existing above and below the focal plane were completely bleached by single-photon excitation, whereas in two-photon excitation, the bleached area was limited to the vicinity of the focal plane.9, 45 Therefore, EGFP fluorescence recovered more rapidly in two-photon FRAP. Fig. 1FRAP analyses of EGFP-histone H1.2. (a) The heterochromatic region within an open box of living HeLa cell nuclei was excited and bleached by single-photon excitation using laser light with a wavelength of . Scale bar indicates . (b) The heterochromatic region within an open box of living HeLa cell nuclei was excited and bleached by two-photon excitation using femtosecond laser light with a wavelength of . Scale bar indicates . (c) Circles and squares indicate fluorescence recovery curve obtained from single- and two-photon FRAP, respectively. 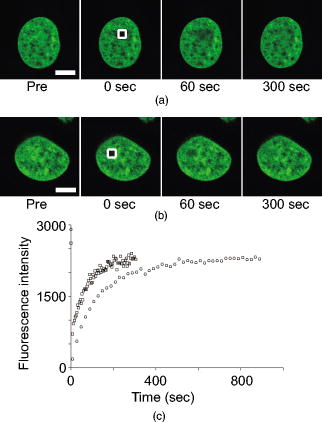 We established a tobacco BY-2 cell line stably expressing histone H1-sGFP (a GFP variant in which threonine is substituted for serine at 65) and analyzed the histone H1 mobility in plant cultured cells by two-photon FRAP (Fig. 2 ). The BY-2 cells have spherical nuclei, which are different from the flat nuclei in mammalian adhesive cultured cells. The recovery rate in two-photon FRAP using a tobacco BY-2 cell was also approximately four times faster than that in single-photon FRAP. This result indicates that two-photon FRAP is also suitable for investigating molecules in thick subcellular organelles. Furthermore, living cells are damaged to a lesser extent when two-photon excitation is used for in-vivo analyses, because a near-infrared laser is employed for the excitation. We compared the recovery rate of GFP-tagged histone H1 between single- and two-photon FRAP in tobacco BY-2 cells, and obtained similar results both in HeLa and tobacco BY-2 cells. We found that the recovery rate is not mainly dependent on the kinds of host cells, but types of target proteins. Fig. 2FRAP analysis of histone H1-sGFP in BY-2 cells. (a) The heterochromatic region of living BY-2 cell nuclei was excited and bleached by two-photon excitation using femtosecond laser light with a wavelength of . Arrow shows the bleaching area. Scale bar indicates . (b) Fluorescence recovery curve obtained from two-photon FRAP.  2.2.Two-Photon Fluorescence Recovery after Photobleaching for Analyzing Histone with Low Mobility In VivoFRAP is an indispensable method for analyzing the molecular dynamics in living cells. Analysis of the dynamics of molecules with low mobility by conventional single-photon FRAP is time consuming, as observed for core histones whose of the slow fraction is more than .42 We also established a HeLa cell line to stably express one of the core histones, namely, histone H2A (Fig. 3 ).9 In the case of histone H2A, the recovery rate in two-photon FRAP has been reported to be twice faster than that in single-photon FRAP.9 As described in the prevous section, in the case of histone H1, the recovery rate can be reduced to approximately one-fourth using two-photon FRAP. This demonstrates that two-photon FRAP is a powerful tool to analyze molecules with low mobility in vivo. 3.Femtosecond Laser Nanosurgery3.1.Difference between Bleaching and Intracellular AblationCell and intracellular surgery based on femtosecond laser pulses is attractive in cell biology because it allows noninvasive intracellular ablation at subdiffraction resolution to be performed within vital cells. 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 A femtosecond laser pulse at higher energies can produce localized energy absorption, and intracellular ablation occurs only in and around the focal volume, allowing site-specific dissection, removal, or disruption of organelles to be carried out. We previously investigated fluorescence recovery after femtosecond laser irradiation to distinguish between bleaching and intracellular ablation.17 Here, we used the EGFP-tagged histone H1 for visualization of the nucleus.44 Figure 4 shows fluorescence images of the EGFP-tagged nucleus before and after femtosecond laser irradiation with different energies (0.26 and /pulse) at a wavelength of . A nuclear region of in a living HeLa cell expressing EGFP-histone H1 was irradiated. Figure 5 shows the relative fluorescence intensity of the bleached area after femtosecond laser irradiation. We defined the relative fluorescence intensity as the fluorescence intensity after femtosecond laser irradiation divided by that before irradiation. At the energy of /pulse, fluorescence disappeared after irradiation and fluorescence recovery was not observed. At the energy of /pulse, fluorescence disappeared after irradiation; however, fluorescence recovery was observed. This result demonstrates that the fluorophore was bleached; however, subsequent recovery of fluorescence in the bleached region occurred due to the exchange of unbleached fluorophore molecules. Fig. 4FRAP analysis of enhanced green fluorescent protein (EGFP)-labeled nucleus at different energies. An EGFP-labeled nuclear region within an open box of in a living HeLa cell was irradiated. Scale bar indicates .  After femtosecond laser irradiation, we restained the cell with Hoechest 33342. The nuclei normally became uniformly stained after the addition of of Hoechest 33342, and therefore, we obtained fluorescence images after . When intracellular ablation of the nucleus was performed at /pulse, no fluorescence was observed from EGFP or Hoechest 33342 in the laser-irradiated region. When bleaching occurred at /pulse, fluorescence was observed from both EGFP and Hoechest 33342. These results confirm that fluorescence recovery is an indicator of intracellular ablation of organelles in living cells. Table 1 summarizes the differences between bleaching and intracellular ablation. Table 1Difference between bleaching and intracellular ablation.
x=not
observed,
o=observed
3.2.Intracellular Ablation of MitochondriaWe previously showed intracellular ablation of individual mitochondria in living HeLa cells using a femtosecond laser.17 Here, we show the viability of the cells after femtosecond laser radiation by use of a femtosecond laser oscillator at a repetition rate of . The cells expressed the enhanced yellow fluorescent protein (EYFP) in the mitochondria. Femtosecond laser pulses with an energy of /pulse were focused on the cells. Figures 6a and 6b show the stacked confocal images obtained before and after irradiation. The figures show that fluorescence from a single mitochondrion disappeared. That is, a mitochondrion irradiated by the femtosecond laser pulses did not exhibit fluorescence recovery. We also confirmed intracellular ablation of the mitochondrion by restaining with MitoTracker Red. Note that displacement of the mitochondria outside the focal region between the before and after images was attributed to cytoplasmic streaming, which indicated the viability of the cells. The viability of the cells after intracellular ablation was also verified by the observation of cell division. These experiments demonstrate that femtosecond laser pulses can be used to ablate a specific organelle from living cells without compromising their viability. Fig. 6Stacked 3-D confocal images (a) before and (b) after femtosecond laser irradiation with /pulse (exposure time: ). Target mitochondrion is indicated by the arrow. Scale bar indicates .  The viability of the cells after femtosecond laser radiation was ascertained by using PI. The membranes of living cells are impermeable to PI; it only penetrates dead or apoptotic cells with compromised membranes. PI was added to the medium just after irradiation of the femtosecond laser pulses, and fluorescence images were observed after . Cells whose mitochondria were ablated at the energy of /pulse were not stained by the PI. The viability of the cells after femtosecond laser irradiation was ascertained by impermeability of PI, as well as by the presence of cytoplasmic streaming. These experiments demonstrate that the cells remained viable when mitochondria were ablated by femtosecond laser pulses. In contrast, at the energy of /pulse, red fluorescence derived from PI was observed in the nuclei, because the plasma membrane was distorted or destroyed by laser irradiation, allowing PI to penetrate the cell (Fig. 7 ), even if the focal point was a mitchondrion beneath the cell membrane. At the energy of /pulse, cytoplasmic streaming in the cells was not observed. To perform intracelluar ablation of mitochondria in a living cell without compromising cell viability, it is important to select optimal laser energy. Fig. 7Confocal image of the cells after femtosecond laser irradiation and the addition of PI. Just after irradiation of femtosecond laser pulses, PI was added to the culture. The fluorescence images were observed after addition of the PI. At an energy of /pulse, red fluorescence derived from PI was observed in the nuclei. Scale bar indicates . 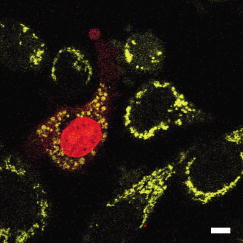 4.Labeling of Single Organelle by Two-Photon ConversionWe have reported photoconversion of a single mitochondrion labeled with Kaede by two-photon excitation.39 In this section, we show the photoconversion of individual mitochondria labeled with Kaede at different sites by two-photon excitation. Femtosecond laser pulses were focused into a target mitochondrion to alter the fluorescence from green to red. Figures 8a and 8b show stacked 3-D confocal fluorescence images obtained before and after femtosecond laser irradiation. We used living BY-2 cells whose mitochondria were labeled with Kaede. Three different mitochondria were sequentially irradiated. After photoconversion by the femtosecond laser irradiation, the red fluorescent signal at laser-irradiated mitochondria increased in intensity. The results demonstrate that mitochondria at specific positions were marked by photoconversion from green to red. The cross sectional spatial resolution and depth resolution of the photoconversion were approximately . Fig. 8Site-specific photoconversion of Kaede within mitochondria in a living BY-2 cell. Stacked 3-D confocal images (a) before and (b) after femtosecond laser pulse irradiation. Individual mitochondria were photoconverted from green to red by femtosecond laser pulses. Laser pulses with an energy of /pulse were focused at the mitochondria indicated by the arrows, and the shutter was opened for an exposure time of . Scale bar indicates . 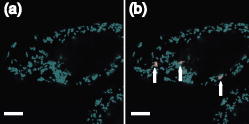 We tracked the movement of the photoconverted mitochondria using the red photoconversion of Kaede. Figures 9a and 9b show confocal images obtained after photoconversion and the trajectories of individual mitochondria after . Four mitochondria were marked by photoconversion. The trajectories were plotted based on time-lapse images at intervals of . The velocities of the photoconverted mitochondria were calculated to be 0.15, 0.38, 0.23, and , respectively. Fig. 9(a) Starting position of four mitochondria after photoconversion. (b)Tracking of particular mitochondria after . The lines of four different colors indicate the tracking of each mitochondrion. The trajectories of the mitochondrion are shown by the lines. Scale bar indicates . 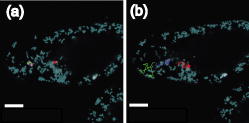 Site-specific organelle marking enabled us to track the dynamics of individual organelles at different sites in a living cell. The results demonstrated the ability to track the dynamics of individual mitochondra and to reveal detailed spatial information in a living cell, such as position and velocity. The ability to label selective organelles is a potentially promising method for analysis of intracellular structures by tracking the dynamics of individual organelles in a living cell, thus revealing the mechanisms involved in organelle dynamics. 5.DiscussionTo date, the mobility properties of some types of proteins have been reported by using conventional single-photon FRAP. However, these data reflect only the 2-D mobility of target proteins; the -axis mobility cannot be monitored directly. Brown previously showed that the diffusion constant could not be exactly calculated by single-photon FRAP.7 Moreover, if the target proteins have only -axis mobility, the results obtained from conventional single-photon FRAP might cause misunderstanding, not only quantitatively but also qualitatively. In contrast, using two-photon FRAP, we can monitor the mobility of the target proteins not only in the and directions but also in the direction.9 We attempted to compare the properties of single- and two-photon FRAP theoretically in terms of kinetics of the molecules. According to Fick’s law, the diffusion rate of molecules in a solution depends on their concentration and the square of the interfacial surface area. When the concentration of GFP-histone H1 in the nucleus is constant, the recovery rate depends on the volume and square of the interface of the bleached area. Our results indicate that, using two-photon FRAP with core histone H2A, the recovery rate can be reduced to approximately one-half. This clearly indicates that two-photon FRAP is suitable for detecting molecules with low mobility. Another advantage of two-photon excitation is less photodamage in living cells. The viability of the Drosophila embryo was dramatically improved by two-photon excitation compared with the case of single-photon excitation.26 Thus, our study clearly shows that two-photon FRAP is a superior method for obtaining accurate data. For example, this method allows analyzing the molecule mobility in the spherical nuclei. The laser power for two-photon excitation was stronger than that for single-photon excitation in bleaching the fluorescence of EGFP-histone H1.2, although the same square region was bleached at the focal plane. This was due to the differences in the cross section between single-photon absorption and two-photon absorption.46 Following two-photon excitation using a laser with such high power, we confirmed the viability of the target cells by Hoechst 33342/PI staining. All cells were found to be viable after the two-photon excitation (data not shown). Intracellular ablation of organelles has been demonstrated using both low-repetition-rate amplified laser systems and high-repetition-rate oscillators. At low repetition rate, amplified laser systems can provide high pulse energy, which causes less thermal damage. At low repetition rate , material modification is produced by a single pulse due to the formation of a high-density plasma. In this regime, the pulse energy is slightly above the threshold for optical breakdown. Intracellular ablation relies on the thermoelastic-induced formation of cavitation bubbles.11 We performed intracellular ablation of mitochondria using 250 pulses at an energy of /pulse from a Ti:sapphire amplifier.13, 14 In contrast, in a high-repetition-rate femtosecond laser system (typically megahertz order), focusing femtosecond laser pulses in a cell results in an increase in temperature in a localized region surrounding the focal spot through heat accumulation, and material modification occurs due to the formation of a low-density plasma and a cumulative effect.11 In this regime, pulse energy is below the optical breakdown threshold for a single pulse. Intracellular ablation is caused by free-electron-induced chemical decomposition.11 In our experiments using a Ti:sapphire oscillator at a repetiton rate of , intracellular ablation of mitochondria was performed using pulses at an energy of /pulse. The energy was lower than that achieved by the amplified laser, and therefore, many pulses were necessary. After intracellular ablation of a mitochondrion in both high and low repetition rate regimes, the viability of the cells after femtosecond laser irradiation was verified by the observation of cell division and by the presence of cytoplasmic streaming. The results demonstrate that the intracellular ablation of an organelle can be performed without compromising cell viability. We have demonstrated tracking the movement of mitochondria by monitoring the photoconverted fluorescence of Kaede. By photoconversion of the fluorescence properties at different specific points by moving the focal position of tightly focused femtosecond laser pulses, the movement of a specific organelle at different sites in a living cell could be tracked. Selective marking techniques of intracellular objects provide spatial and temporal information about intracellular objects and their dynamics within a living cell. 6.Methods and Materials6.1.Optical SetupFigure 10 shows a schematic diagram of the setup used for imaging of fluorescence-labeled organelles in living cells using continuous wave (cw) lasers and manipulation of organelles using a femtosecond laser oscillator. We used a confocal microscope to image the fluorescence. The laser scanning microscope was adapted from an Olympus FV300 scanning unit (Olympus Corp., Tokyo, Japan) combined with an Olympus IX71 inverted microscope. The cw beams from a He-Ne laser (wavelength ) and an laser (wavelength ) were reflected by dichroic mirrors DM2 and DM3 and then focused into a BY-2 cell through an oil-immersion objective lens (OB, Olympus, , NA 1.4). Fluorescence was excited by the cw laser light, and the backpropagating fluorescence was collected using the same objective lens and detected with photomultiplier tubes, after passing through a bandpass filter. 2-D confocal crosssectional images were obtained by scanning the focused laser beams in the plane with a pair of high-speed galvanometer mirrors (GM) inside the laser-scanning microscope. Scanning in the depth direction ( direction) was achieved by moving the objective lens with a stepping motor to obtain 3-D confocal fluorescence images. To construct the stacked 3-D images, we obtained 13 confocal crosssectional images by translating the objective lens by in the depth direction in steps of . Imaging and marking by laser irradiation were performed using Fluoview software. 6.2.Tobacco BY-2 CellsHistone H1-sGFP-expressing tobacco BY-2 cells were prepared according to the previously reported procedure.47 Kaede-expressing tobacco BY-2 cells were prepared by a previously described method,37 and the cells were maintained as previously described by Nagata, Nenoto, and Hasezawa.48 The cells were cultured in modified Linsmaier and Skoog medium in a rotary shaker at in the dark. Two-day-old BY-2 cells, after subculturing, were used for our analyses. 6.3.HeLa CellsHeLa human carcinoma cells were cultured and maintained as previously reported.49 For visualization of the chromosomes, the HeLa cells were transfected by pEGFP-C1/histone H1.2 using LipofectAMINE plus reagent (Invitrogen Corp., Calsbad, California).44 Fusion constructs containing a gene encoding enhanced yellow fluorescent protein EYFP and the mitochondrial targeting sequence from human cytochrome c oxidase (pEYFP-Mito, Clonetech) were transfected into HeLa cells using the calcium phosphate method. In the restaining experiments, mitochondria of the HeLa cells were stained with MitoTracker Red CMH2XRos(chloramethyl-X-rosamine, Molecular Probes), which is a mitochondria-selective dye, for at . To determine the effects on cell viability after femtosecond-laser irradiation, PI (Sigma, St. Louis, Missouri) was added to the medium after the irradiation. 6.4.Single-Photon and Two-Photon Fluorescence Recovery after PhotobleachingHeLa cells were transfected with the constructed vectors using Lipofectamine (Invitrogen) or FuGENE6 (Roche Diagnostics, Basel, Switzerland) following the corresponding manufacturer’s protocols. Transfectants were selected with of G418 (Sigma) and cloned by the limiting dilution method as described previously.7 HeLa cells were inoculated onto glass-bottomed dishes (Matsunami Glass, Inc., Kishiwada, Japan), maintained in Dulbecco’s modified Eagle’s medium (DMEM) without phenol-red (Invitrogen) with 2-[4-(2-hydroxyethyl)-1-piperadinyl] ethansulfonic acid (HEPES, pH 7.2) in a humidified atmosphere on a heated stage, and were imaged using an inverted fluorescence microscope (IX-71, Olympus) with a objective lens. Single-photon FRAP was performed using an FV300 confocal microscopic system ( laser, wavelength ). To carry out single-photon FRAP, the entire image was taken using illumination from the laser at a mean power of ( at the sample) before the microscope, and then a nuclear region of was bleached, while scanning with the galvanometer mirrors, by increasing the power of the laser to ( at the sample). After , subsequent single-photon fluorescence images were captured with the original settings. Two-photon FRAP with femtosecond laser pulses was performed using a mode-locked Ti:sapphire laser oscillator with a wavelength of and a repetition rate of (Coherent, Mira, Santa Clara, California). The laser pulses passed through a series of SF10 prisms to compensate for the dispersion of the optical components in the light path and the microscope. The entire imaging was performed using two-photon excitation at a wavelength of with a mean power of ( at the sample). A nuclear region of was bleached at a power of ( at the sample) by scanning with the galvanometer mirrors. After bleaching, subsequent images were captured with the original settings. 6.5.Intracellular AblationIntracellular ablation of mitochondria was performed using a mode-locked Ti:sapphire laser oscillator with a wavelength of and a repetition rate of (Coherent, Mira). To perform ablation in the nuclear region, an EGFP-labeled nuclear region in a living HeLa cell was irradiated at a wavelength of . To obtain fluorescence images of GFP and YFP, bandpass filters BP1 (transmission wavelength: ) and BP2 (transmission wavelength: ) were placed before photomultiplier tubes PMT1 and PMT2, respectively. The number of pulses supplied was selected by an electromagnetic shutter (Sigma Koki, -65L, Tokyo, Japan). 6.6.Photoconversion of KaedePhotoconversion of Kaede was performed using near-infrared femtosecond laser pulses from a mode-locked Ti:sapphire laser oscillator with a wavelength of . The green fluorescence was obtained using a single-photon fluorescence microscope. laser light passed through a bandpass filter BP1 (transmission wavelength: ) and the green fluorescence was detected with a photomultiplier tube (PMT1). Red fluorescence after photoconversion of Kaede was excited by the He-Ne laser light (wavelength ) and detected with a photomultiplier tube (PMT2), after passing through a bandpass filter BP2 (transmission wavelength: ). 7.ConclusionsIn summary, we describe site-specific photoconversion, FRAP analysis, and ablation of intracellular organelles by two-photon excitation. Intracellular imaging of a specific organelle in combination with optical manipulation, such as intracellular ablation and FRAP, will help in understanding the mechanisms and interactions of intracellular organelles. AcknowledgmentsThe authors thank T. Shimada, K. Isobe, N. Arakawa, H. Ishii, A. Morimoto, and D. Kurihara from Osaka University for assistance in the experiments. The authors would like to thank N. Tsutsumi and S. Arimura from the University of Tokyo for the preparation of Kaede. Part of this work on intracellular ablation was supported by JSPS and DFG under the German Research Cooperative Program (Watanabe). This work was supported by BIRD of Japan Science and Technology Agency and KAKENHI from MEXT of Japan (Matsunaga). ReferencesW. Denk,
J. H. Strickler, and
W. W. Webb,
“Two-photon laser scanning fluorescence microscopy,”
Science, 248
(4951), 73
–76
(1990). https://doi.org/10.1126/science.2321027 0036-8075 Google Scholar
C. Xu,
W. Zipfel,
J. B. Shear,
R. M. Williams, and
W. W. Webb,
“Multiphoton fluorescence excitation: New spectral windows for biological nonlinear microscopy,”
Proc. Natl. Acad. Sci. U.S.A., 93
(20), 10763
–10768
(1996). https://doi.org/10.1073/pnas.93.20.10763 0027-8424 Google Scholar
K. König,
“Multiphoton microscopy in life science,”
J. Microsc., 200
(2), 83
–104
(2000). https://doi.org/10.1046/j.1365-2818.2000.00738.x 0022-2720 Google Scholar
S. Coscoy,
F. Waharte,
A. Gautreau,
M. Martin,
D. Louvard,
P. Mangreat,
M. Arpin, and
F. Amblard,
“Molecular analysis of microscopic ezrin dynamics by two-photon FRAP,”
Proc. Natl. Acad. Sci. U.S.A., 99
(20), 12813
–12818
(2002). 0027-8424 Google Scholar
F. Waharte,
C. M. Brown,
S. Coscoy,
E. Coudrier, and
F. Amblard,
“A two-photon FRAP analysis of the cytoskeleton dynamics in the microvilli of intestinal cells,”
Biophys. J., 88
(2), 1467
–1478
(2005). 0006-3495 Google Scholar
E. Van Keuren and
W. Schrof,
“Fluorescence recovery after two-photon bleaching for the study of dye diffusion in polymer systems,”
Macromolecules, 36
(13), 5002
–5007
(2003). 0024-9297 Google Scholar
E. B. Brown,
E. S. Wu,
W. Zipfel, and
W. W. Webb,
“Measurement of molecular diffusion in solution by multiphoton fluorescence photobleaching recovery,”
Biophys. J., 77
(5), 2837
–2849
(1999). 0006-3495 Google Scholar
R. Peters,
J. Peters,
K. H. Tews, and
W. Bahr,
“A microfluorimetric study of translational diffusion in erythrocyte,”
Biochim. Biophys. Acta, 367 282
–294
(1974). https://doi.org/10.1016/0005-2736(74)90085-6 0006-3002 Google Scholar
T. Higashi,
S. Matsunaga,
K. Isobe,
A. Morimoto,
T. Shimada,
S. Kataoka,
W. Watanabe,
S. Uchiyama,
K. Itoh, and
K. Fukui,
“Histone H2A mobility is regulated by its N- and C-terminal tails, and acetylation of core histone tails,”
Biochem. Biophys. Res. Commun., 57
(3), 627
–632
(2007). 0006-291X Google Scholar
M. F. Yanik,
H. Cinar,
H. N. Cinar,
A. D. Chisholm,
Y. Jin, and
A. Ben-Yakar,
“Functional regeneration after laser axotomy,”
Nature (London), 432
(7019), 822
–822
(2004). https://doi.org/10.1038/432822a 0028-0836 Google Scholar
A. Vogel,
J. Noack,
G. Hüttman, and
G. Paltauf,
“Mechanisms of femtosecond laser nanosurgery of cells and tissues,”
Appl. Phys. B, 81
(8), 1015
–1047
(2005). https://doi.org/10.1007/s00340-005-2036-6 0946-2171 Google Scholar
U. K. Tirlapur and
K. König,
“Femtosecond near infrared lasers as a novel tool for non-invasive real-time high resolution time-lapse imaging of chloroplast division in living bundle sheath cells of Arabidopsis,”
Planta, 214
(1), 1
–10
(2001). 0032-0935 Google Scholar
W. Watanabe,
N. Arakawa,
S. Matsunaga,
T. Higashi,
K. Fukui,
K. Isobe, and
K. Itoh,
“Femtosecond laser disruption of subcellular organelles in a living cell,”
Opt. Express, 12
(18), 4203
–4213
(2004). https://doi.org/10.1364/OPEX.12.004203 1094-4087 Google Scholar
W. Watanabe,
S. Matsunaga,
T. Shimada,
T. Higashi,
K. Fukui, and
K. Itoh,
“Femtosecond laser disruption of mitochondria in living cells,”
Med. Laser Appl., 20
(3), 185
–191
(2005). 1615-1615 Google Scholar
N. Shen,
D. Datta,
C. B. Schaffer,
P. LeDuc,
D. E. Ingber, and
E. Mazur,
“Ablation of cytoskeletal filaments and mitochondria in live cells using a femtosecond laser nanoscissor,”
Mech. Chem. Biosyst., 2
(1), 17
–25
(2005). 1546-2048 Google Scholar
A. Heisterkamp,
I. Z. Maxwell,
E. Mazur,
J. M. Underwood,
J. A. Nickerson,
S. Kumar, and
D. E. Ingber,
“Pulse energy dependence of subcellular dissection by femtosecond laser pulses,”
Opt. Express, 13
(10), 3690
–3696
(2005). https://doi.org/10.1364/OPEX.13.003690 1094-4087 Google Scholar
T. Shimada,
W. Watanabe,
S. Matsunaga,
T. Higashi,
H. Ishii,
K. Fukui,
K. Isobe, and
K. Itoh,
“Intracellular disruption of mitochondria in living HeLa cells with a femtosecond laser oscillator,”
Opt. Express, 13
(24), 9869
–9880
(2005). https://doi.org/10.1364/OPEX.13.009869 1094-4087 Google Scholar
K. König,
“Laser tweezers and multiphoton microscopes in life sciences,”
Histochem. Cell Biol., 114
(2), 79
–92
(2000). 0948-6143 Google Scholar
K. König,
I. Riemann, and
W. Fritzsche,
“Nanodissection of human chromosomes with near-infrared femtosecond laser pulses,”
Opt. Lett., 26
(11), 819
–821
(2001). https://doi.org/10.1364/OL.26.000819 0146-9592 Google Scholar
U. K. Tirlapur and
K. König,
“Targeted transfection by femtosecond laser,”
Nature (London), 418
(6895), 290
–291
(2002). https://doi.org/10.1038/418290a 0028-0836 Google Scholar
W. Supatto,
D. Débarre,
B. Moulia,
E. Brouzés,
J. L. Martin,
E. Farge, and
E. Beaurepaire,
“In vivo modulation of morphogenetic movements in Drosophila embryos with femtosecond laser pulses,”
Proc. Natl. Acad. Sci. U.S.A., 102
(4), 1047
–1052
(2005). https://doi.org/10.1073/pnas.0405316102 0027-8424 Google Scholar
L. Sacconi,
I. M. Tolić-Nørrelykke,
R. Antolini, and
F. S. Pavone,
“Combined intracellular three-dimensional imaging and selective nanosurgery by a nonlinear microscope,”
J. Biomed. Opt., 10
(1), 014002
(2005). https://doi.org/10.1117/1.1854675 1083-3668 Google Scholar
N. M. Wakida,
C. S. Lee,
E. T. Botvinick,
L. Z. Shi,
A. Dvornikov, and
M. W. Berns,
“Laser nanosurgery of single microtubules reveals location-dependent depolymerization rates,”
J. Biomed. Opt., 12
(2), 024022
(2007). https://doi.org/10.1117/1.2718920 1083-3668 Google Scholar
J. Lippincott-Schwartz and
G. H. Patterson,
“Development and use of fluorescent protein markers in living cells,”
Science, 300
(5616), 87
–91
(2003). https://doi.org/10.1126/science.1082520 0036-8075 Google Scholar
G. H. Patterson and
J. Lippincott-Schwartz,
“A photoactivatable GFP for selective photolabeling of proteins and cells,”
Science, 297
(5588), 1873
–1877
(2002). https://doi.org/10.1126/science.1074952 0036-8075 Google Scholar
J. N. Post,
K. A. Lidke,
B. Rieger, and
D. J. Arndt-Jovin,
“One- and two-photon photoactivation of a paGFP-fusion protein in live Drosophila embryos,”
FEBS Lett., 579
(2), 325
–330
(2005). https://doi.org/10.1016/j.febslet.2004.11.092 0014-5793 Google Scholar
M. Karbowski,
D. Arnoult,
H. Chen,
D. C. Chan,
C. L. Smith, and
R. J. Youle,
“Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis,”
J. Cell Biol., 164
(4), 493
–499
(2004). https://doi.org/10.1083/jcb.200309082 0021-9525 Google Scholar
M. Schneider,
S. Barozzi,
I. Testa,
M. Faretta, and
A. Diaspro,
“Two-photon activation and excitation properties of PA-GFP in the region,”
Biophys. J., 89
(2), 1346
–1352
(2005). https://doi.org/10.1529/biophysj.104.054502 0006-3495 Google Scholar
D. M. Chudakov,
V. V. Belousov,
A. G. Zaraisky,
V. V. Novoselov,
D. B. Staroverov,
D. B. Zorov,
S. Lukyanov, and
K. A. Lukyanov,
“Kindling fluorescent proteins for precise in vivo photolabeling,”
Nat. Biotechnol., 21
(2), 191
–194
(2003). https://doi.org/10.1038/nbt778 1087-0156 Google Scholar
D. M. Chudakov,
V. V. Verkhusha,
D. B. Staroverov,
E. A. Souslova,
S. Lukyanov, and
K. A. Lukyanov,
“Photoswitchable cyan fluorescent protein for protein tracking,”
Nat. Biotechnol., 22
(11), 1435
–1439
(2004). https://doi.org/10.1038/nbt1025 1087-0156 Google Scholar
Y. Chen,
P. J. Macdonald,
J. P. Skinner,
G. H. Patterson, and
J. D. Müller,
“Probing nucleocytoplasmic transport by two-photon activation of PA-GFP,”
Microsc. Res. Tech., 69
(3), 220
–226
(2006). 1059-910X Google Scholar
J. Wiedenmann,
S. Ivanchenko,
F. Oswald,
F. Schmitt,
C. Rocker,
A. Salih,
K. D. Spindler, and
G. U. Nienhaus,
“EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion,”
Proc. Natl. Acad. Sci. U.S.A., 101
(45), 15905
–15910
(2004). https://doi.org/10.1073/pnas.0403668101 0027-8424 Google Scholar
N. G. Gurskaya,
V. V. Verkhusha,
A. S. Shcheglov,
D. B. Staroverov,
T. V. Chepurnykh,
A. F. Fradkov,
S. Lukyanov, and
K. A. Lukyanov,
“Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light,”
Nat. Biotechnol., 24
(4), 461
–465
(2006). https://doi.org/10.1038/nbt1191 1087-0156 Google Scholar
R. Ando,
H. Hama,
M. Yamamoto-Hino,
H. Mizuno, and
A. Miyawaki,
“An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein,”
Proc. Natl. Acad. Sci. U.S.A., 99
(20), 12651
–12656
(2002). https://doi.org/10.1073/pnas.202320599 0027-8424 Google Scholar
H. Mizuno,
T. Kumar Mal,
K. I. Tong,
R. Ando,
T. Furuta,
M. Ikura, and
A. Miyawaki,
“Photo-induced peptide cleavage in the green-to-red conversion of a fluorescent protein,”
Mol. Cell, 12
(4), 1051
–1058
(2003). 1097-2765 Google Scholar
H. Tsutsui,
S. Karasawa,
H. Shimizu,
N. Nukina, and
A. Miyawaki,
“Semi-rational engineering of a coral fluorescent protein into an efficient highlighter,”
EMBO Rep., 6
(3), 233
–238
(2005). 1469-221X Google Scholar
S. Arimura,
J. Yamamoto,
G. P. Aida,
M. Nakazono, and
N. Tsutsumi,
“Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution,”
Proc. Natl. Acad. Sci. U.S.A., 101
(20), 7805
–7808
(2004). https://doi.org/10.1073/pnas.0401077101 0027-8424 Google Scholar
S. Ivanchenko,
C. Röcker,
F. Oswald,
J. Wiedenmann, and
U. Nienhaus,
“Targeted green-red photoconversion of EosFP, a fluorescent marker protein,”
J. Biol. Phys., 31
(3–4), 249
–259
(2005). https://doi.org/10.1007/s10867-005-0174-z 0092-0606 Google Scholar
W. Watanabe,
T. Shimada,
S. Matsunaga,
D. Kurihara,
S. Arimura,
N. Tsutsumi,
K. Fukui,
K. Isobe, and
K. Itoh,
“Single organelle tracking by two-photon conversion,”
Opt. Express, 15
(5), 2490
–2498
(2007). https://doi.org/10.1364/OE.15.002490 1094-4087 Google Scholar
T. Misteli,
A. Gunjan,
R. Hock,
M. Bustin, and
D. T. Brown,
“Dynamic binding of histone H1 to chromatin in living cells,”
Nature (London), 408
(6814), 877
–881
(2000). 0028-0836 Google Scholar
M. A. Lever,
J. P. H. Th’ng,
X. J. Sun, and
M. J. Hendzel,
“Rapid exchange of histone H1.1 on chromatin in living human cells,”
Nature (London), 408
(6814), 873
–876
(2000). https://doi.org/10.1038/35048603 0028-0836 Google Scholar
H. Kimura and
P. R. Cook,
“Kinetics of core histones in living human cells: Little exchange of H3 and H4 and some rapid exchange of H2B,”
J. Biol. Phys., 153
(7), 1341
–1353
(2001). 0092-0606 Google Scholar
D. Y. Chen,
M. Dundr,
C. Wang,
A. Leung,
A. Lamond,
T. Misteli, and
S. Huang,
“Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins,”
J. Biol. Phys., 168
(1), 41
–54
(2005). 0092-0606 Google Scholar
A. E. Gambe,
R. M. Ono,
S. Matsunaga,
N. Kutsuna,
T. Higaki,
T. Higashi,
S. Hasezawa,
S. Uchiyama, and
K. Fukui,
“Development of a multistage classifier for a monitoring system of cell activity based on imaging of chromosomal dynamics,”
Cytometry, 71A
(5), 286
–296
(2007). 0196-4763 Google Scholar
W. R. Zipfel,
R. M. Williams, and
W. W. Webb,
“Nolineear magic: multiphoton microscopy in the biosciences,”
Nat. Biotechnol., 21
(11), 1369
–1377
(2003). https://doi.org/10.1038/nbt899 1087-0156 Google Scholar
J. P. Hermann and
J. Ducuing,
“Absolute measurement of two-photon cross sections,”
Phys. Rev. A, 5
(6), 2557
–2568
(1972). https://doi.org/10.1103/PhysRevA.5.2557 1050-2947 Google Scholar
A. Kawabe,
S. Matsunaga,
K. Nakagawa,
D. Kurihara,
A. Yoneda,
S. Hasezawa,
S. Uchiyama, and
K. Fukui,
“Characterization of plant Aurora kinases during mitosis,”
Plant Mol. Biol., 58
(1), 1
–13
(2005). 0167-4412 Google Scholar
T. Nagata,
Y. Nemoto, and
S. Hasezawa,
“Tobacco BY-2 cell-line as the Hela-cell in the cell biology of higher-plants,”
Intl. Rev. Cytol. Survey Cell Biol., 132 1
–30
(1992). Google Scholar
T. Higashi,
E. Nagamori,
T. Sone,
S. Matsunaga, and
K. Fukui,
“A novel transfection method for mammalian cells using calcium alginate microbeads,”
J. Biosci. Bioeng., 97
(3), 191
–195
(2004). 1389-1723 Google Scholar
|