|
|
1.IntroductionThe angiotensin (Ang) type I receptor , a member of the superfamily of G protein-coupled receptors (GPCRs), is important in the regulation of cardiovascular function.1 Similar to other GPCRs,2 on agonist activation, recruits adaptor proteins with clathrin to form stable clathrin-coated vesicles.1 , in clathrin-coated vesicles, fuse with sorting endosomes shortly after internalization.3 This process and additional intracellular trafficking of are thought to be regulated by several Rab GTPases.4, 5, 6, 7, 8 To date, over 60 mammalian Rab proteins have been identified and each Rab protein appears to associate with particular subcellular compartment(s).9, 10, 11, 12, 13 Rab5, located in the cytoplasmic side of the plasma membrane in coated vesicle pits and early endosome membranes, regulates the internalization and sorting of GPCRs, including -adrenergic, and endothelin A and B receptors.4, 9 It is believed that are sorted to Rab5-positive early endosomes after internalization.3 Glutathione S-transferase (GST)-tagged Rab5 has been reported to coimmunoprecipitate with , even in the basal state; however, the colocalization of with Rab5 has not been shown by confocal microscopy.5, 6 Rab7, predominantly localized in large vesicles and late endocytic compartments, has been suggested to regulate vesicle trafficking between late endosomes and lysosomes,10 particularly in the vacuolar protein sorting toward and away from lysosomes.11, 12 However, the mechanisms underlying the Rab5/7-regulated endocytic trafficking are not well understood. Recently, cytoskeletons have been reported to be important in the Rab GTPase-mediated regulation of endocytic trafficking.7, 14 As in yeast,15 the actin cytoskeleton has been reported to be important in the formation of clathrin-coated pits and the regulation of the initial steps of internalization in mammalian cells.1, 7, 14 The microtubule cytoskeleton has also been suggested to be involved in the regulation of endocytic trafficking.16 However, the precise roles of actin and microtubule cytoskeletons in the intracellular trafficking remain to be determined. Even though the has been reported to be targeted to lysosomes for degradation,17, 18 the rat , overexpressed in human embryonic kidney (HEK) 293 cells, has not been shown to localize in late endosomes and lysosomes following Ang II stimulation.5, 6 Therefore, the role of Rab5 and Rab7 in the cellular trafficking of , including its lysosomal degradation, is not clear. The limited resolution of confocal microscopy used in previous studies provides the rationale for additional studies, using methods that can localize associations between cellular compartments with much higher resolution [e.g., fluorescence lifetime imaging–Förster/fluorescence resonance energy transfer (FLIM-FRET) microscopy]. FLIM-FRET techniques are increasingly applied in biological and biomedical research because of their high sensitivity in detecting protein-protein interactions over the range of .19, 20, 21 Utilizing these techniques, we reported recently that the is degraded in proteasomes on stimulation of the dopamine receptor in human renal proximal tubule and HEK293 cells.22 This is in contrast to the lysosomal degradation of on binding with its endogenous ligand, Ang II.17, 18 However, the dynamic regulation of degradation following Ang II stimulation is not well understood. In this report, in HEK293 cells heterologously expressing the human tagged with enhanced green fluorescent protein (EGFP) as the donor and Alexa 555-labeled Rab5, Rab7, and LAMP1 as the acceptors, we used conventional confocal microscopy, multiphoton FLIM-FRET microscopy, and biochemical studies to determine the roles of Rab5 and Rab7, as well as actin and microtubule cytoskeletons, in the dynamic endocytic trafficking of . We found that following Ang II treatment, the was internalized into early endosomes, regulated by Rab5 while the trafficking to late endosomes for eventual degradation was regulated by Rab7. Latrunculin A, an actin polymerization inhibitor,23 but not nocodazole, a microtubule polymerization inhibitor,24 blocked the Ang II-induced FRET occurrence between -EGFP and Rab5 or Rab7, labeled with Alexa 555, indicating the requirement of intact actin but not microtubule cytoskeleton in the regulation of endocytic trafficking by these Rab proteins in these HEK293 cells. The occurrence of FRET between -EGFP and LAMP1-Alexa 555 following Ang II stimulation was impaired by photobleaching the acceptor, confirming the trafficking of into lysosomes, detected by FLIM-FRET. These data demonstrate that Rab5/7-regulated trafficking for eventual lysosomal degradation, following Ang II stimulation is actin cytoskeleton dependent. 2.Materials and Methods2.1.Antibodies and ReagentsMonoclonal mouse Rab5 antibodies were purchased from BD Transduction Laboratories (Lexington, Kentucky); polyclonal rabbit anti-Rab5, polyclonal rabbit anti-Rab7, polyclonal rabbit anti-GFP, and normal mouse and rabbit antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, California); polyclonal rabbit LAMP1 antibody was from Affinity Bioreagents, Inc. (Rockford, Illinois); monoclonal GFP antibody was purchased from Clontech (Mountain View, California); Alexa Fluor 546 anti-mouse, Alexa Fluor 633 anti-rabbit, Alexa Fluor 633 anti-mouse secondary antibodies, Alexa Fluor 546 phalloidin, and Alexa-555 protein labeling kits were purchased from Invitrogen (Eugene, Oregon). Monoclonal -tubulin antibody and Ang II were purchased from Sigma (St. Louis, Missouri). Latrunculin A and nocodazole were purchased from Calbiochem (Gibbstown, New Jersey). 2.2.Cell Culture and TransfectionHEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Gaithersburg, Maryland), containing glucose, 10% fetal bovine serum, L-glutamine, and sodium pyruvate. Human -EGFP, or its empty vector pEGFP-N1,25 was transfected into HEK293 cells using Lipofectamine™ 2000 transfection reagents (Invitrogen), according to the manufacturer’s instructions, as previously described.26 2.3.Immunoprecipitation and ImmunoblottingHEK293 and -HEK293 cells were sonicated in lysis buffer ( Tris⋅HCl, pH sucrose), with benzamidine hydrochloride and protease inhibitor cocktail (Thermo Scientific, Rockford, Illinois). The homogenates were incubated (rocking, , ) with of anti-GFP rabbit IgG, of anti-Rab5 rabbit IgG, or of anti-Rab7 rabbit IgG in of the lysis buffer with , 0.1% ovalbumin, 4-(2-aminoethyl)benzene sulfonyl fluoride (AEBSF), 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. Normal rabbit IgG was used as negative control. After the addition of of a 50% slurry of protein G-Sepharose CL-4B (Amersham Pharmacia, Piscataway, New Jersey) in phosphate-buffered saline (PBS) and incubation at overnight, the beads were washed three times with of ice-cold PBS, containing AEBSF. Proteins bound to beads were eluted and separated, as previously described.22 2.4.Confocal Immunofluorescence MicroscopyCells on coverslips were fixed with 4% paraformaldehyde in PBS for at room temperature. After washing with PBS, the fixed cells were incubated overnight at with Rab5 mAb, anti-Rab7 rabbit IgG, or anti-LAMP1 rabbit IgG. After washing, the coverslips were incubated with Alexa Fluor or secondary antibodies for at . Coverslips were mounted in SlowFade mounting medium (Invitrogen, Eugene, Oregon) and sealed onto glass slides. Samples were imaged using an Olympus Fluoview FV300 laser scanning confocal microscope, equipped with a oil-immersion objective. Quantitative analysis was performed using MetaMorph 6.1 (Molecular Devices, Downingtown, Pennsylvania). Background was subtracted from each image and then the images were thresholded to identify specific protein fluorescence. Percentage of overlap of integrated intensity measurement was determined for over Rab5, Rab7, or LAMP1. 2.5.FLIM SystemThe FLIM system was described in our previous report.27 Briefly, we used a Nikon TE300 epi-fluorescence microscope equipped with a Plan Fluor water-immersion IR objective. The microscope was coupled to a Biorad Radiance 2100 confocal/multiphoton system with the LaserSharp2000 software. A Verdi pumped, tunable mode-locked ultrafast pulsed laser (Mira 900, Coherent, Inc., Santa Clara, California) was tuned to the wavelength to excite the donor fluorophore (EGFP). Emitted photons were collected by a fast photomultiplier tube with a bandpass emission filter. The lifetime photomultiplier tube was coupled in the connection arm of the external detector with a response time of (PMC-100-0, Becker & Hickl GmbH, Berlin, Germany). The use of the time-correlated single-photon-counting module board (SPC-150, Becker & Hickl GmbH, Berlin, Germany) with a minimum temporal resolution of allowed pixel-by-pixel registration of the accumulated photons. A fluorescence decay histogram of photons at different emission times relative to the laser excitation pulse was generated from the distribution of interpulse intervals at each pixel of the image. 2.6.FLIM Data AcquisitionPolyclonal rabbit Rab5, Rab7, and LAMP1 IgG were digested by papain and separated by Sephadex G-100, and then conjugated with Alexa 555, according to the manufacturer’s instructions (Alexa Fluor 555 protein labeling kits, Invitrogen, Eugene, Oregon). -HEK293 cells were treated with vehicle, Ang II ( , 5 and ) without or with pretreatment with latrunculin A , an actin polymerization inhibitor23 or nocodazole , a microtubule polymerization inhibitor24 and fixed with 4% paraformaldehyde in PBS for at room temperature. After washing with PBS, cells on coverslips were incubated overnight at with Alexa Fluor 555-conjugated Fab fragment (with a 3:1 of dye: Fab ratio) of polyclonal rabbit Rab5, Rab7, or LAMP1. Donor fluorophores were continuously scanned at the wavelength with appropriate excitation average power for to accumulate enough photon counts for fluorescence lifetime analysis. The same settings were used for samples in the presence or absence of the acceptor fluorophores (Alexa 555-labeled Rab5, Rab7, or LAMP1). In additional experiments, the DPSS laser, equipped on a ZEISS 510 Meta microscope system, was employed to photobleach the acceptor fluorophores (at least 50% intensity) in the region of interest or whole visual fields. A series of pre- and postbleaching FLIM data sets were acquired using the FLIM system, as described above. The comparison of the -EGFP lifetimes obtained before and after photobleaching the acceptor fluorophores was determined by Student’s t-test from 74–89 cells from three independent sets of experiments. 2.7.FLIM Data AnalysisThe data analysis was performed using the SPCImage software (V2.9, Becker & Hickl GmbH, Berlin, Germany), which also provides an estimate of the instrument response function of the FLIM system. The fluorescence lifetimes of the donor fluorophores were obtained with single or double exponential fitting on a pixel-by-pixel basis based on a weighted least-squares numerical approach as previously described.20, 27 The unquenched donor lifetime was obtained by fitting the donor-alone data with the single exponential decay. The double exponential fittings were employed to extract the quenched lifetime of the donor fluorophores, in the presence of the acceptor fluorophores. The fitting significance was judged based on the value and visually comparing the fitting curve with the raw data points. The average lifetime is used for comparison of a change in lifetime. 3.Results3.1.Cellular Localization of Human and Rab ProteinsConsistent with previous observations,18, 26 tagged with EGFP was observed primarily at the plasma membrane, but also in the intracellular compartments and cytoplasm in HEK293 cells, under basal conditions [Fig. 1 ]. Endogenous Rab5 was observed in the cytoplasm as punctuate vesicles of different sizes and intracellular membrane networks; slight staining of the plasma membrane was also observed [Fig. 1]. Endogenous Rab7 was scattered throughout the cytoplasm in punctuate vesicles of varying sizes, some of which were observed in perinuclear areas [Fig. 1]. Minimal colocalization was observed between and nuclei [Fig. 1]. However, some colocalization between and Rab5 [Fig. 1], and between and Rab7 [Fig. 1] was observed (see Fig. 3 for quantification). Fig. 1Subcellular distributions of (a) , (b) Rab5, and (c) Rab7 in -HEK293 cells. The , tagged with EGFP at its C-terminus, is localized mainly at the plasma membrane. Both Rab5-Alexa 546 and Rab7-Alexa 633 are seen as punctuate structures throughout the cytoplasm. (d) overlay of with DAPI (blue) nuclear stain, (e) overlay of (a) ( -EGFP, green) and (b) (Rab5-Alexa 546, red), and (f) overlay of (a) and (c) (Rab7-Alexa 633, blue). Bar, .  Fig. 3Quantitative analysis of the colocalizations of -EGFP with Rab5-Alexa 546, Rab7-Alexa 633, and LAMP1-Alexa 633 in -HEK293 cells. -HEK293 cells were treated with vehicle (basal) or with Ang II for 5 or , as indicated. Quantitative analysis was carried out using MetaMorph 6.1 (Molecular Device, Downingtown, Pennsylvania), as described in section 2. Data are , cells per group. ANOVA, Student-Newman-Keuls test. ∗, versus Basal; #, versus Ang II treatment.  As shown in Fig. 2 , , as expected, internalized on activation with Ang II . Endocytosis was more obvious with the than with the treatment [Figs. 2 and 2]. Ang II treatment did not induce any obvious change in the subcellular localization of Rab5 [Fig. 2 versus Fig. 1] and Rab7 [Fig. 2 versus Fig. 1]. However, the relationship between and these Rab proteins was changed dramatically compared to the basal state (Fig. 3 ). Colocalization was observed between and Rab5 at [Fig. 2]. At this time point, the colocalization between and Rab7 and between and LAMP1 was modest [Figs. 2, 3, 7], close to the basal state [Figs. 1 and 3]. When the duration of the Ang II treatment was increased to , was observed to colocalize with both Rab5 [Figs. 2 and 3] and Rab7 [Figs. 2 and 3]. There were a few vesicles showing the colocalization of the three proteins [Fig. 2]. Fig. 2Internalization of induced by Ang II treatment for (a–f) and (g–l) in -HEK293 cells. Ang II induced the cellular internalization of (a,g) , but not (b,h) Rab5 or (c,i) Rab7. (d,j) overlays of -EGFP (green) and Rab5-Alexa 546 (red); (e,k) overlays of -EGFP (green) and Rab7-Alexa 633 (blue), and (f,l) overlays of -EGFP (green), Rab5-Alexa 546 (red), and Rab7-Alexa 633 (blue). Bar, . 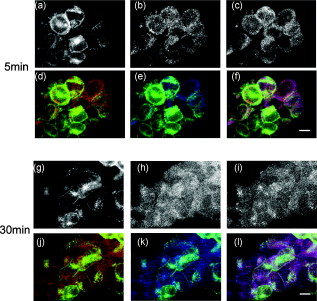 Fig. 7Subcellular distribution of -EGFP and LAMP1-Alexa 633 in -HEK293 cells. Cells were treated with Ang II , at the indicated time periods, as described in Section 2. Green, -EGFP; Red, LAMP1-Alexa 633; yellow, colocalization. Bar, . (Color online only.) 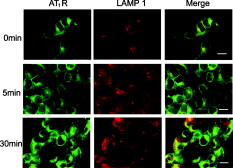 3.2.Coimmunoprecipitation of with Rab ProteinsIn agreement with the morphological observations, both Rab5 [Fig. 4 ] and Rab7 [Fig. 4] coimmunoprecipitated with GFP antibody after of Ang II treatment, that became more obvious after of treatment (Fig. 4). (-EGFP) also coimmunoprecipitated with Rab5 and Rab7 antibodies with Ang II treatment (data not shown). There was no coimmunoprecipitation in the basal state, which is consistent with the absence of obvious colocalization between and Rab5 or Rab7 observed with confocal microscopy (Fig. 1), but not with a previous report using GST-tagged Rab5 in transiently transfected COS7 cells.6 Whether or not the discrepancy is caused by the tagging of Rab528 or cell-specific trafficking remains to be determined. Fig. 4Coimmunoprecipitation of -EGFP with (a) Rab5 and (b) Rab7 in -HEK293 cells. Cells were treated with Ang II , at the indicated periods. One mg of cell lysates were incubated with anti-GFP IgG at . The precipitated protein complexes were immunoblotted with (a) Rab5 mAb or (b) anti-Rab7 IgG. Both Rab5 and Rab7 coimmunoprecipitated with -GFP in the samples treated with Ang II for and . NR, normal rabbit IgG; LC, IgG light chain; NS, nonspecific band; Lys, whole cell lysate.  3.3.Dynamic Interaction of with Rab ProteinsTo investigate further the interaction between and Rab proteins, FLIM-FRET analyses were performed in -HEK293 cells with -EGFP, as the donor fluorophore, and Rab5 or Rab7 Fab conjugated Alexa Fluor 555, as the acceptor fluorophores. First, we measured the fluorescence lifetime of -EGFP, in absence of any acceptor. Without Ang II treatment, the average lifetime was [Fig. 5, 5, 5 ]. In the absence of any acceptor, Ang II treatment, for or [Fig. 5, 5, 5], did not change that lifetime (Table 1 ). We also measured the lifetime of -EGFP in the presence of Fab of Alexa Fluor 555 that was not conjugated to Rab proteins. Additional controls included EGFP, as the donor, in the presence or absence of Alexa Fluor 555, as the acceptor. The EGFP lifetimes obtained in all these controls were similar (Table 1). Fig. 5Analysis of the -EGFP lifetime in the absence of acceptor in -HEK293 cells. To separate the images from the background, the threshold was set at 50 (the minimum number of photon counts in the peak). The histograms (b,e) show the distribution of lifetime images of -EGFP (a,d). The decay graphs (c,f) show the trace of the fit (red) to the photon decay data (blue) at a particular pixel in the lifetime image. Scale bar, . (Color online only.)  Table 1Lifetimes of AT1R -EGFP in AT1R -HEK293 cells. Cells were treated with Ang II (100nM) at the indicated time periods and fixed, as described in Section 2. N , number of cells analyzed; data are mean±S.E. ; ANOVA, Student–Newman–Keuls test. No significant differences among the control groups were detected.
Next, we studied the lifetime of -EGFP in the presence of Rab proteins conjugated with Fab of Alexa Fluor 555. In the basal state, -EGFP lifetime was not changed by the presence of Rab7-Alexa 555, but slightly decreased in the presence of Rab5-Alexa 555 (Table 2 ), compared to that measured from the donor-alone control samples (Table 1). Treatment with Ang II for shortened the -EGFP lifetime in the presence of Rab5-Alexa 555, but not in the presence of Rab7-Alexa 555, relative to their corresponding values at basal state (Table 2), indicating FRET occurrence between -EGFP with Rab5-Alexa 555, but not with Rab7-Alexa 555. When the Ang II treatment was increased to , the -EGFP lifetime was shortened in the presence of both Rab5 and Rab7 labeled with Alexa 555 (Table 2), indicating FRET occurrence between and these two Alexa 555-labeled Rab proteins. Table 2Effect of Ang II on the lifetime of AT1R -EGFP in AT1R -HEK293 cells. Cells were treated with Ang II (100nM) at the indicated time periods and fixed, as described in Section 2. Data are mean±S.E. ; N , number of cells analyzed; ANOVA, Student-Newman-Keuls test.
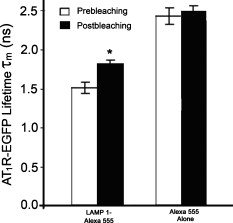
3.4.Cytoskeleton-Dependent Interactions with Rab ProteinsNext, we studied the role of the cytoskeleton in the interaction of with Rab5 or Rab7 by cytoskeleton polymerization inhibitors. Latrunculin A, a blocker of the polymerization of actin filaments [Fig. 6 ] but not microtubules [Fig. 6], did not affect -EGFP lifetime in the presence of Rab5- or Rab7-Alexa 555 without Ang II treatment (Table 2). However, latrunculin A pretreatment blocked the ability of Ang II to shorten the -EGFP lifetime in the presence of Rab5- or Rab7-Alexa 555 (Table 2), indicating the requirement of intact actin for Rab5 and Rab7 to regulate intracellular trafficking. Nocodazole, which disrupts the polymerization of microtubules [Fig. 6] but not actin [Fig. 6], also had no significant effect on -EGFP lifetime on samples not treated with Ang II, in the presence of Rab5- or Rab7-Alexa 555 (Table 2). However, in contrast to latrunculin A pretreatment, nocodazole pretreatment did not block the ability of Ang II to shorten the -EGFP lifetime, in the presence of Rab5- or Rab7-Alexa 555. The -EGFP lifetimes measured after Ang II treatment with and without nocodazole pretreatment were similar (Table 2), indicating that intact microtubules are not required in the Rab5/7-mediated intracellular trafficking of . Fig. 6Cytoskeletal disruption with cytoskeleton inhibitors in -HEK293 cells. Cells were treated with (a,b) vehicle (DMSO), (c,d) latrunculin A, or (e,f) nocodozale, and stained with fluorescein phalloidin to visualize F-actin (a,c,e) and anti-tubulin to visualize microtubules (b,d,f), as described in Section 2. Scale bar, .  3.5.FLIM-FRET Analysis of Association with LysosomesFollowing stimulation with Ang II, has been proposed to traffic to Rab7-positive late endosomes for eventual degradation in lysosomes.1, 3, 4 Therefore, we next investigated the relationship between and LAMP1, a marker protein for lysosomes. Confocal microscopy showed the colocalization of with LAMP1 following a Ang II treatment, but only modest colocalization with a Ang II treatment, close to the basal state (Figs. 3 and 7 ). To corroborate the morphological studies, FLIM-FRET experiments were performed to observe the -EGFP lifetime, in the presence of LAMP1-Alexa 555. Similar to the basal state with Rab5-Alexa 555, there was a modest decrease in -EGFP lifetime in the presence of LAMP1-Alexa 555 [Fig. 8 ], compared to that obtained with the donor-alone samples. The decrease in -EGFP lifetime in the basal state could have been caused by the acidic environment of lysosomes (see Section 4). With Ang II treatment, no obvious lifetime change was observed [Fig. 8], compared to its basal state, indicating no FRET occurrence. The Ang II treatment resulted in a dramatic decrease in the lifetime of -EGFP, in some locations, indicating FRET occurrence between -EGFP and LAMP1-Alexa 555 in those locations [Fig. 8], suggesting this portion of degraded in lysosomes. Chloroquine, a lysosome inhibitor, abrogated the lifetime change that occurred with the Ang II treatment (data not shown). Fig. 8FLIM-FRET analysis of -EGFP interaction with LAMP1-Alexa 555 in -HEK293 cells. Cells were treated with (a) vehicle ( cells), Ang II for (b) ( cells) or (c) ( cells), as described in Section 2. The -EGFP lifetime was analyzed in the presence of LAMP1-Alexa 555. Scale bar, . 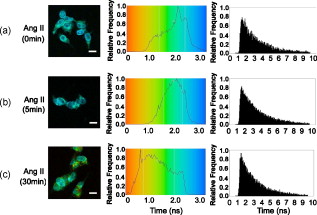 To confirm the observation of FRET occurrence between -EGFP and LAMP1-Alexa 555 with the Ang II treatment detected by FLIM-FRET analysis, acceptor photobleaching experiments were performed. The average lifetime of -EGFP was ( cells) after photobleaching the acceptor, which was significantly longer than that obtained prior to bleaching ( , cells). In contrast, photobleaching the EGFP alone-Alexa 555 alone samples by the DPSS 561 laser (using the same power and duration of photobleaching) did not change the donor lifetime (Fig. 9 ). These studies support the conclusion of an association of with LAMP1 and that following Ang II stimulation, the is trafficked to lysosomes for eventual degradation. 4.Discussion, similar to other GPCRs, undergoes a series of intracellular trafficking processes on stimulation with its agonists. The sorting endosome is the first compartment in the intracellular trafficking pathway in which is targeted to its destination, either for recycling to the plasma membrane or to the late endosome for eventual lysosomal degradation.3, 4 Rab5 is considered important in the early endosome fusion.6, 9 Although the GTPase activity of Rab5 does not regulate the internalization of ,3, 5 it interacts with the C-terminal domain of in a yeast two-hybrid system.6 Our current study, showing the colocalization (Fig. 2), and coimmunoprecipitation (Fig. 4) of Rab5 with , as well as the shortening of lifetime (Table 2) following Ang II treatment, is consistent with a role of Rab5 in the vesicle fusion and sorting of to early endosomes.3, 5, 6 The trafficking of GPCRs from sorting endosomes to late endosomes is not as well established as their fusion with early sorting endosomes.14 Rab7 is speculated to regulate this process because of its localization in late endosomes and lysosomes.10 Overexpression of Rab7 and its active form Rab7-Q67L, but not its dominant-negative form Rab7-N125I, has been reported to increase the lysosomal degradation of in response to Ang II treatment.5 Our current study, showing the colocalization and coimmunoprecipitation of Rab7 with (Figs. 2 and 4), as well as the shortening of lifetime (Table 2) following Ang II treatment, is consistent with a role of Rab7 in the sorting of to late endosomes. The molecular mechanisms involved in Rab5- and Rab7-regulated GPCR endocytic trafficking and sorting pathways are not well known.4, 5, 11, 14 Both actin and microtubule cytoskeletons play important roles in the regulation of Rab GTPase-mediated intracellular trafficking of membrane proteins.4, 11, 29, 30 Some GPCRs (e.g., parathyroid hormone receptor) mainly require actin cytoskeleton for their intracellular trafficking;31 other GPCRs (e.g., adenosine receptor) mainly require microtubules;32 while some other GPCRs (e.g., metabotropic glutamate receptor 5) require both actin and microtubules for their intracellular trafficking.33 It has been established that the actin cytoskeleton is important in the internalization and trafficking of through early and late endosomes or recycling endosomes following the formation of clathrin-coated vesicles upon Ang II stimulation.1, 3, 7, 8, 14 Recently, a role of microtubules in the regulation of signaling and intracellular trafficking has been demonstrated. Kolb 34 reported that the microtubule is important in the externalization (apical recycling) of . In rat renal proximal tubule cells, disruption of microtubules inhibits apical Ang II-induced generation,35 indicating that microtubules are needed for function in apical but not in basolateral membrane. Li 36, 37 have also reported that microtubules are important in the trafficking after of Ang II treatment. However, they suggested that the initial steps in membrane endocytosis may involve clathrin-coated pits, presumably via actin-dependent mechanisms. Our current study, using FLIM-FRET microscopy and cytoskeleton inhibitors, provides direct evidence for a role of actin in the early stages of intracellular trafficking following Ang II stimulation. Both actins and microtubules are probably important in the trafficking that is time (early versus late) and location (apical versus basolateral membrane) specific. Actins provide forces for membrane invaginations, vesicle propelling, scalfolding protein-protein interaction, and modulation of membrane plasticity,29 while microtubules are mainly responsible for the movement of newly budded vesicles and important for directed transport, particularly in polarized cells.30 In agreement with Li, 36, 37 microtubules are not important in the initial steps of intracellular trafficking upon Ang II stimulation (Table 2). Instead, intact actin motors are utilized in lieu of microtubules in the early trafficking of . The utility of conventional confocal microscopy in evaluating protein-protein interaction is limited by its resolution . This barrier can be overcome by using FRET microscopy, which provides a sensitive tool for investigating a variety of biological phenomena that produce changes in molecular proximity over a range of . However, most of intensity-based FRET techniques require complicated algorithms to remove the spectral bleed-through from both donor and acceptor fluorophores.38 In contrast, FLIM-FRET microscopy allows quantitative monitoring of protein-protein interactions by measuring the change in lifetime of the donor fluorophore, EGFP in the current experiments. In FLIM-FRET microscopy, spectral bleed-through is usually not an issue because only the donor signals are monitored, and fluorescence lifetime is not influenced by probe concentration, excitation light intensity, light scattering, or stoichiometry; these characteristics make FLIM-FRET microscopy an accurate method for FRET measurements.20, 21, 27 FLIM-FRET microscopy was used in the current study to observe the change of -EGFP lifetime, in the presence of fluorescently labeled Rab5, Rab7, and LAMP1. Over the last decade, various pairs of mutant green fluorescent protein (GFP) fluorophores have been used successfully in FLIM-FRET studies.19, 20, 21 In our system, the donor is the EGFP-tagged and the acceptor is the Alexa 555-labeled Fab fragment of Rab5, Rab7, or LAMP1. Cy3-labeled antibody, as the acceptor, paired with GFP, as the donor, has been used previously in COS7 cells.39 The ability of antibody-labeled acceptor fluorophores to increase the chance of ideal orientation with the donor EGFP, and the easier application of antibody labeling are advantages over the double DNA plasmid transfection method. In this study, the lifetime of -EGFP is shortened slightly but significantly, in the presence of Alexa fluor 555-labeled Rab5 and LAMP1, relative to that in the donor-alone samples in the basal state (Table 2). With Ang II stimulation, the lifetime of -EGFP is decreased markedly (Table 2). The decrease in -EGFP lifetime could be due to the interaction of with these acceptors or a change in the local environment around the molecules, such as pH, concentration of and other ions.19, 20, 21 The acidic environment can also cause a conformational change of donor and acceptor fluorophores. The luminal pH is supposed to decrease in sorting and late endosomes, and especially in lysosomes.40 The decrease in -EGFP lifetime in the basal state could be due to the acidic pH of sorting endosomes and lysosomes.19, 20, 21 However, the acidic pH is not the only explanation because the same phenomenon is not observed in the presence of Rab7-Alexa 555 (Table 2). Fluorescence lifetime can be affected by many local environment factors, such as pH, concentration of , and other ions.41, 42 Nonetheless, we may still be able to interpret the FLIM-FRET results properly because they are corroborated by both morphological and biochemical studies. The characterization of pH and other local environmental factors on their effect on -EGFP lifetime remains to be studied. We have reported previously that the is sorted into proteasomes from the plasma membrane through the interaction with following the addition of fenoldopam, a agonist.22 The lifetime of (-EGFP, the donor) is shortened to with stimulation, in the presence of ubiquitin as the acceptor fluorophore, suggesting that a fraction of is polyubiquitinated and processed for proteasomal degradation.22 Our previous FRET studies showed that some of the internalized escape degradation and recycle back to the plasma membrane, coordinately, via Rab4 and Rab11 following Ang II stimulation.26 In this study, Ang II treatment shortens the -EGFP lifetime, in the presence of Rab5, Rab7, and LAMP1 labeled with Alexa 555 (Table 2, Figs. 8 and 9), in a time-coordinated manner. These results indicate that internalized are sorted into Rab5-positive sorting endosomes, subsequently into Rab7-positive late endosomes, and then to lysosomes for eventual degradation. It is not clear how cells control these distinct endocytic pathways for in different situations. Rab7 has been suggested recently as a potential master switch that controls distinct intracellular endocytic pathways through its role in the recruitment of retromer complexes proteins, Rab-interacting proteins, and cytoskeletons.12 Whether or not this is the case for endocytic trafficking requires further investigation. AcknowledgmentsWe are grateful to Dr. G. Guillemette (Université de Sherbrooke, Canada) for the -GFP construct and its empty vector pEGFP-N1, and Dr. Susette Mueller (Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC) for the assistance with confocal microscopy studies. This study was supported in part by the W. M. Keck Foundation, University of Virginia, and grants from the National Institutes of Health, No. HL074940, No. HL23081, No. DK39308, No. HL68686, and No. HL092196. ReferencesM. de Gasparo,
K. J. Catt,
T. Inagami,
J. W. Wright, and
T. Unger,
“International union of pharmacology. XXIII. The angiotensin II receptors,”
Pharmacol. Rev., 52 415
–472
(2000). 0031-6997 Google Scholar
P. A. Jose,
G. M. Eisner, and
R. A. Felder,
“Renal dopamine receptors in health and hypertension,”
Pharmacol. Ther., 80 149
–182
(1998). https://doi.org/10.1016/S0163-7258(98)00027-8 0163-7258 Google Scholar
C. Oro,
H. Qian, and
W. G. Thomas,
“Type 1 angiotensin receptor pharmacology: signaling beyond G proteins,”
Pharmacol. Ther., 113 210
–226
(2007). https://doi.org/10.1016/j.pharmthera.2006.10.001 0163-7258 Google Scholar
A. Marchese,
M. M. Paing,
B. R. Temple, and
J. Trejo,
“G protein-coupled receptor sorting to endosomes and lysosomes,”
Annu. Rev. Pharmacol. Toxicol., 48 601
–629
(2008). https://doi.org/10.1146/annurev.pharmtox.48.113006.094646 0362-1642 Google Scholar
L. B. Dale,
J. L. Seachrist,
A. V. Babwah, and
S. S. Ferguson,
“Regulation of angiotensin II type 1A receptor intracellular retention, degradation, and recycling by Rab5, Rab7, and Rab11 GTPases,”
J. Biol. Chem., 279 13110
–13118
(2004). https://doi.org/10.1074/jbc.M313333200 0021-9258 Google Scholar
J. L. Seachrist,
S. A. Laporte,
L. B. Dale,
A. V. Babwah,
M. G. Caron,
P. H. Anborgh, and
S. S. Ferguson,
“Rab5 association with the angiotensin II type 1A receptor promotes Rab5 GTP binding and vesicular fusion,”
J. Biol. Chem., 277 679
–685
(2002). https://doi.org/10.1074/jbc.M109022200 0021-9258 Google Scholar
P. K. Mehta and
K. K. Griendling,
“Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system,”
Am. J. Physiol.: Cell Physiol., 292 C82
–97
(2007). https://doi.org/10.1152/ajpcell.00287.2006 0363-6143 Google Scholar
Z. Gáborik and
L. Hunyady,
“Intracellular trafficking of hormone receptors,”
Trends Endocrinol. Metab., 15 286
–293
(2004). https://doi.org/10.1016/j.tem.2004.06.009 Google Scholar
C. Bucci,
R. G. Parton,
I. H. Mather,
H. Stunnenberg,
K. Simons,
B. Hoflack, and
M. Zerial,
“The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway,”
Cell, 70 715
–728
(1992). https://doi.org/10.1016/0092-8674(92)90306-W 0092-8674 Google Scholar
Y. Feng,
B. Press, and
A. Wandinger-Ness,
“Rab7: an important regulator of late endocytic membrane traffic,”
J. Cell Biol., 131 1435
–1452
(1995). https://doi.org/10.1083/jcb.131.6.1435 0021-9525 Google Scholar
S. Pfeffer,
“Membrane domains in the secretory and endocytic pathways,”
Cell, 112 507
–517
(2003). https://doi.org/10.1016/S0092-8674(03)00118-1 0092-8674 Google Scholar
R. Rojas,
T. van Vlijmen,
G. A. Mardones,
Y. Prabhu,
A. L. Rojas,
S. Mohammed,
A. J. Heck,
G. Raposo,
P. van der Sluijs, and
J. S. Bonifacino,
“Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7,”
J. Cell Biol., 183 513
–526
(2008). https://doi.org/10.1083/jcb.200804048 0021-9525 Google Scholar
P. Del Conte-Zerial,
L. Brusch,
J. C. Rink,
C. Collinet,
Y. Kalaidzidis,
M. Zerial, and
A. Deutsch,
“Membrane identity and GTPase cascades regulated by toggle and cut-out switches,”
Mol. Syst. Biol., 4 206
(2008). 1744-4292 Google Scholar
A. Sorkin and
M. von Zastrow,
“Endocytosis and signalling: intertwining molecular networks,”
Nat. Rev. Mol. Cell Biol., 10 609
–622
(2009). https://doi.org/10.1038/nrm2748 1471-0072 Google Scholar
M. Kaksonen,
C. P. Toret, and
D. G. Drubin,
“A modular design for the clathrin- and actin-mediated endocytosis machinery,”
Cell, 123 305
–320
(2005). https://doi.org/10.1016/j.cell.2005.09.024 0092-8674 Google Scholar
L. Zuo,
M. Ushio-Fukai,
L. L. Hilenski, and
R. W. Alexander,
“Microtubules regulate angiotensin II type 1 receptor and Rac1 localization in caveolae/lipid rafts: role in redox signaling,”
Arterioscler., Thromb., Vasc. Biol., 24 1223
–1228
(2004). https://doi.org/10.1161/01.ATV.0000132400.25045.2a 1079-5642 Google Scholar
L. Hein,
L. Meinel,
R. E. Pratt,
V. J. Dzau, and
B. K. Kobilka,
“Intracellular trafficking of angiotensin II and its and receptors: evidence for selective sorting of receptor and ligand,”
Mol. Endocrinol., 11 1266
–1277
(1997). https://doi.org/10.1210/me.11.9.1266 0888-8809 Google Scholar
L. Hunyady,
A. J. Baukal,
Z. Gaborik,
J. A. Olivares-Reyes,
M. Bor,
M. Szaszak,
R. Lodge,
K. J. Catt, and
T. Balla,
“Differential PI 3-kinase dependence of early and late phases of recycling of the internalized AT1 angiotensin receptor,”
J. Cell Biol., 157 1211
–1222
(2002). https://doi.org/10.1083/jcb.200111013 0021-9525 Google Scholar
I. A. Demarco,
A. Periasamy,
C. F. Booker, and
R. N. Day,
“Monitoring dynamic protein interactions with photoquenching FRET,”
Nat. Methods, 3 519
–524
(2006). https://doi.org/10.1038/nmeth889 1548-7091 Google Scholar
H. Wallrabe and
A. Periasamy,
“Imaging protein molecules using FRET and FLIM microscopy,”
Curr. Opin. Biotechnol., 16 19
–27
(2005). https://doi.org/10.1016/j.copbio.2004.12.002 0958-1669 Google Scholar
A. Periasamy and
R. M. Clegg,
“FLIM applications in the biomedical sciences,”
FLIM Microscopy in Biology and Medicine, 385
–400 Chapman & Hall/CRC, Boca Raton
(2009). Google Scholar
H. Li,
I. Armando,
P. Yu,
C. Escano,
S. C. Mueller,
L. Asico,
A. Pascua,
Q. Lu,
X. Wang,
V. A. Villar,
J. E. Jones,
Z. Wang,
A. Periasamy,
Y. S. Lau,
P. Soares-da-Silva,
K. Creswell,
G. Guillemette,
D. R. Sibley,
G. Eisner,
J. J. Gildea,
R. A. Felder, and
P. A. Jose,
“Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells,”
J. Clin. Invest., 118 2180
–2189
(2008). https://doi.org/10.1172/JCI33637C1 0021-9738 Google Scholar
M. Coué,
S. L. Brenner,
I. Spector, and
E. D. Korn,
“Inhibition of actin polymerization by latrunculin A,”
FEBS Lett., 213 316
–318
(1987). https://doi.org/10.1016/0014-5793(87)81513-2 0014-5793 Google Scholar
M. Clarke,
J. Köhler,
J. Heuser, and
G. Gerisch,
“Endosome fusion and microtubule-based dynamics in the early endocytic pathway of dictyostelium,”
Traffic, 3 791
–800
(2002). https://doi.org/10.1034/j.1600-0854.2002.31104.x Google Scholar
P. M. Lanctot,
P. C. Leclerc,
M. Clément,
M. Auger-Messier,
E. Escher,
R. Leduc, and
G. Guillemette,
“Importance of N-glycosylation positioning for cell-surface expression, targeting, affinity and quality control of the human receptor,”
Biochem. J., 390 367
–376
(2005). https://doi.org/10.1042/BJ20050189 0264-6021 Google Scholar
H. Li,
H. F. Li,
R. A. Felder,
A. Periasamy, and
P. A. Jose,
“Rab4 and Rab11 coordinately regulate the recycling of angiotensin II type I receptor as demonstrated by fluorescence resonance energy transfer microscopy,”
J. Biomed. Opt., 13 031206
(2008). https://doi.org/10.1117/1.2943286 1083-3668 Google Scholar
Y. Chen and
A. Periasamy,
“Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization,”
Microsc. Res. Tech., 63 72
–80
(2004). https://doi.org/10.1002/jemt.10430 1059-910X Google Scholar
R. Rozenfeld and
L. A. Devi,
“Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3,”
FASEB J., 22 2311
–2322
(2008). https://doi.org/10.1096/fj.07-102731 0892-6638 Google Scholar
L. Lanzetti,
“Actin in membrane trafficking,”
Curr. Opin. Cell Biol., 19 453
–458
(2007). https://doi.org/10.1016/j.ceb.2007.04.017 0955-0674 Google Scholar
J. P. Caviston and
E. L. Holzbaur,
“Microtubule motors at the intersection of trafficking and transport,”
Trends Cell Biol., 16 530
–537
(2006). https://doi.org/10.1016/j.tcb.2006.08.002 0962-8924 Google Scholar
B. R. Conway,
L. K. Minor,
J. Z. Xu,
M. R. D’Andrea,
R. N. Ghosh, and
K. T. Demarest,
“Quantitative analysis of agonist-dependent parathyroid hormone receptor trafficking in whole cells using a functional green fluorescent protein conjugate,”
J. Cell Physiol., 189 341
–355
(2001). https://doi.org/10.1002/jcp.10028 0021-9541 Google Scholar
C. Saunders and
L. E. Limbird,
“Disruption of microtubules reveals two independent apical targeting mechanisms for G-protein-coupled receptors in polarized renal epithelial cells,”
J. Biol. Chem., 272 19035
–19045
(1997). https://doi.org/10.1074/jbc.272.30.19035 0021-9258 Google Scholar
A. Serge,
L. Fourgeaud,
A. Hemar, and
D. Choquet,
“Active surface transport of metabotropic glutamate receptors through binding to microtubules and actin flow,”
J. Cell. Sci., 116 5015
–5022
(2003). https://doi.org/10.1242/jcs.00822 0021-9533 Google Scholar
R. J. Kolb,
P. G. Woost, and
U. Hopfer,
“Membrane trafficking of angiotensin receptor type-1 and mechanochemical signal transduction in proximal tubule cells,”
Hypertension, 44 352
–359
(2004). https://doi.org/10.1161/01.HYP.0000136645.90116.1a 0194-911X Google Scholar
J. R. Schelling,
A. S. Hanson,
R. Marzec, and
S. L. Linas,
“Cytoskeleton-dependent endocytosis is required for apical type 1 angiotensin II receptor-mediated phospholipase C activation in cultured rat proximal tubule cells,”
J. Clin. Invest., 90 2472
–2480
(1992). https://doi.org/10.1172/JCI116139 0021-9738 Google Scholar
X. C. Li,
O. A. Carretero,
L. G. Navar, and
J. L. Zhou,
“AT1 receptor-mediated accumulation of extracellular angiotensin II in proximal tubule cells: role of cytoskeleton microtubules and tyrosine phosphatases,”
Am. J. Physiol. Renal Physiol., 291 F375
–383
(2006). https://doi.org/10.1152/ajprenal.00405.2005 Google Scholar
X. C. Li,
U. Hopfer, and
J. L. Zhou,
“ receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway,”
Am. J. Physiol. Renal Physiol., 297 F1342
–F1352
(2009). https://doi.org/10.1152/ajprenal.90734.2008 Google Scholar
Y. Chen and
A. Periasamy,
“Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei,”
J. Fluoresc., 16 95
–104
(2006). https://doi.org/10.1007/s10895-005-0024-1 1053-0509 Google Scholar
T. Ng,
A. Squire,
G. Hansra,
F. Bornancin,
C. Prevostel,
A. Hanby,
W. Harris,
D. Barnes,
S. Schmidt,
H. Mellor,
P. I. H. Bastiaens, and
Peter J. Parker,
“Imaging protein kinase activation in cells,”
Science, 283 2085
–2089
(1999). https://doi.org/10.1126/science.283.5410.2085 0036-8075 Google Scholar
J. R. Casey,
S. Grinstein, and
J. Orlowski,
“Sensors and regulators of intracellular pH,”
Nat. Rev. Mol. Cell Biol., 11 50
–61
(2010). https://doi.org/10.1038/nrm2820 1471-0072 Google Scholar
A. A. Heikal,
S. T. Hess, and
W. W. Webb,
“Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent protein (EGFP): acid-base specificity,”
Chem. Phys., 274 37
–55
(2001). https://doi.org/10.1016/S0301-0104(01)00486-4 0301-0104 Google Scholar
S. Modi,
S. M. G.,
D. Goswami,
G. D. Gupta,
S. Mayor, and
Y. Krishnan,
“A DNA nanomachine that maps spatial and temporal pH changes inside living cells,”
Nat. Nanotechnol., 4 325
–330
(2009). https://doi.org/10.1038/nnano.2009.83 1748-3387 Google Scholar
|