
|
|
1.IntroductionLike other epithelial neoplasms, oral cancer, is driven by the clonal evolution and expansion of abnormal cells across the mucosa, physically apparent as a heterogeneous group of lesions in which appearance is not obviously linked to the genetics of the underlying tissue. Successful management of the disease is dependent upon the detection and treatment of the earlier stages of the disease. However, early detection relies heavily on the clinician’s ability to discriminate sometimes subtle alterations associated with premalignant lesions and cancers from reactive and inflammatory conditions that represent the majority of mucosal abnormalities. This can be challenging, even among experienced oral specialists. Wide field-of-view direct fluorescence visualization (FV) devices have been introduced to enhance the recognition of at-risk tissue.1 These devices utilize blue-violet (400 to 450 nm) excitation light, and detection of 460 nm and longer emission light to visualize tissue autofluorescence. The selective long-pass filter in the eyepiece allows the viewer to directly visualize the pale green autofluorescence that is given off by normal tissue. Tissue fluorescence appears altered (reduced in intensity and altered in color) in areas of dysplastic or cancerous tissue such that abnormal or suspicious tissue shows decreased levels of normal autofluorescence, appearing as a dark brown to black region by comparison to the surrounding healthy tissue. While these devices improve the detection of abnormal tissue, inflammation, infection, and trauma can mimic changes associated with premalignant/cancerous tissue. This situation may be improved by optimizing the way in which light interacts with lesions to extract the maximum amount of biologically relevant information.2,3 An alternate strategy is to directly assess the cells involved in the clonal expansion process through analysis of individual cell nuclear phenotypic changes using automated image analysis. The DNA content of quantitatively stained nuclei has been widely used to identify gross chromosomal alterations (larger than 150 Megabases) in many organ sites associated with neoplasia.4 We have previously reported that the organization of DNA within nuclei is predictive of progression of dysplasia to cancer5,6 and highly correlated with other indices of genetic alteration, such as measured loss of heterozygosity (LOH) and array comparative genomic hybridization.7 The combination of DNA content and subtle alterations in DNA organization have been used to improve the detection of cervical and bronchial cancers and precancers from cytological samples and has demonstrated clinical utility. The objective of this study was to develop and assess a high throughput automated approach based on a combination of subtle alterations in cell DNA amount and organization for the recognition of high-risk oral lesions as detected by direct FV and white light. 2.Materials and Methods2.1.Subjects and Sample CollectionThis study included patients in three groups: 1. patients with severe dysplasia, carcinoma in situ (CIS) and squamous cell carcinoma (SCC) lesions (denoted abnormal set), 2. patients with normal mucosa (denoted normal set), and 3. patients with confounding lesions (denoted confounder set). The abnormal set involved participants in the ongoing NIH/NIDCR-funded oral cancer prediction longitudinal (OCPL) study at the British Columbia Cancer Agency in Vancouver, Canada.8 Normal control samples and confounders came from community clinics of the Oral Mucosal Disease Program at the University of British Columbia. All samples were collected between November 2004 and November 2008 through the application of a stiff brush rubbed against the lesion in the oral cavity (brushing). The study was approved by the Institutional Review Board of the BC Cancer Agency and University of British Columbia. Informed consent was obtained from all subjects. A total of 369 cytological samples (from 369 individuals) were analyzed: (1) 148 samples from pathology-proven sites of SCC, CIS or severe dysplasia (denoted abnormal set); (2) 77 samples from sites with inflammation, infection, or trauma, either biopsy-proven or clinically confirmed as such by an Oral Pathology Specialist in lesions at a three-month follow-up examination (denoted confounder set); and (3) 144 samples from normal sites (denoted normal set). In a conventional dental practice, the prevalence of abnormal lesions is quite low () in most settings,9 the clinician performing the brushing may not always recognize the area of most severe change and may consequently not brush exactly the same area an experienced oral specialist would target. In the cervical screening programs the sample is frequently (5% to 20%) not taken from the targeted area at most risk of being transformed.10,11 Thus, to more realistically represent these conditions, approximately 1 cm of clinically normal oral surface around the lesion as well as the lesion itself was intentionally brushed to reduce the dependency of the system on perfectly targeted brushing samples. Exfoliated cells were collected by brushing the mucosal surface with a soft interdental brush (Innovatek cytology brush 4201-CB8B, The Stevens Company), which had been manually curved at the end prior to use. After 10 to 15 strokes of the brush, the collected cells and the brush were put into a 1.5 ml cryovial containing 800 ul of Preservcyte (Hologic Inc.) and stored at 4 ºC until analysis. 2.2.QTP Imaging System for Quantitative CytologyThe oral brushings were cytospun down onto slides and stained with a modified Feulgen-Thionin reaction,12 which quantitatively stains DNA such that the light absorbed at each location in the nucleus is directly proportional to the amount of DNA at that location. Stained slides were scanned using a modified version of the Cyto-Savant automated quantitative system (Integrative Oncology, BC Cancer Agency) currently used for cervical and sputum screening.13 The software used in the Cyto-Savant system is specifically designed for fully automatic slide scanning and collection of correctly segmented and focused images of all objects on the slide (stained nuclei and debris).13,14 Essentially this is a fully automated microscopy system, which is capable of loading individual slides from a slide box, automatically finding and focusing individual objects on the slide, acquiring images of the objects found, and segmenting objects into nuclei-like objects. The system calculates features for each object, passes these features through a multi-level binary classification tree to recognize and differentiate well-segmented, in focus single epithelial cells from overlapping cells, cell clusters, debris, white blood cells, etc. to finally end up with a collection of epithelial cell images and data for each slide. The imaging system performance characteristics follow the recommendation of the European Society of Analytical Cellular Pathology for ploidy analysis.15 A strict quality control process involving measurements of standard targets, power measurements, and illumination stability evaluated using statistical quality control processes is implemented to ensure the linearity and repeatability of the system for each analysis.16 The system used an illumination wavelength () corresponding to the absorption peak of the Thionin stain. The effective pixel sampling within the plane of the sample was 0.34 μm and the effective pixel sampling area was μm2. Each object (cell or debris) scanned on a slide had 110 features calculated and stored. Each object was subjected to a cell recognition algorithm (originally trained for cervical cell recognition)17 to differentiate cells from debris with a subsequent quick review by an experienced cytotechnologist to ensure that only intact well-focused single cells were used in all subsequent analyses. The review typically takes less than 5 min to perform per slide as the automated algorithms are accurate.17 Various subsets of these cell and slide data were used to generate the algorithms described in this study. 2.3.Selection of Training SetSamples were divided into both training and test sets. The training set consisted of a random selection of 120 of the 144 normal samples and 109 of the 148 abnormal samples in addition to all 77 of the confounder samples. The remaining 24 normal samples and 39 abnormal samples were set aside as the test set to evaluate the performance of the generated algorithms. A random selection of cells from the over 175,000 cells making up all the cells in only the normal and abnormal samples in the training set was used to train the cell by cell classifiers and to determine the thresholds for the frequency of cells displaying characteristics associated with the abnormal samples. These thresholds were then applied to all the cells in both the training and test samples. 2.4.Data AnalysisThe system measures two major aspects of all the cells it detects, the amount of DNA within a cell’s nucleus and how that DNA is distributed within the nucleus.18 A cell’s DNA index is a measure of the amount of chromosomal material (total DNA) within the cell: a value of 1 indicates the normal complement of DNA (46 chromosomes in a G0/G1 cell, or 2.9 billion base pairs); a value of 2 indicates twice the usual amount of DNA or 5.8 billion base pairs. It should be noted that a value of 1 does not necessarily indicate a normal complement of chromosomes, only the presence of billion base pairs. Also the uncertainty in the measurement (, or Mega bases in an imaging cytometry system meeting the guidelines of the ESACP)15 means that almost a full chromosome could be missing without being detected. Cell data in the training set were divided into boxes within discrete DNA index ranges: less than 0.9, 0.9 to 1.1, 1.1 to 1.35, 1.35 to 1.6, 1.6 to 1.95, 1.95 to 2.15, and 2.15 and higher. These DNA content ranges are a superset of those used by Bradley et al.19 and are similar to those used in earlier studies for the classification and grouping of cells in cervical smears and sputum samples.20,21 The second aspect measured by the system involved features related to how the DNA was distributed within the nucleus, frequently referred to as nuclear texture analysis. These measures capture genetic and epigenetic alterations, which are not manifest as large base pair gains or losses but result in changes in the spatial arrangement and packaging of chromosomal strands within the nucleus. To recognize which and how large these measured spatial arrangement changes are needed to differentiate between normal and abnormal cells, for each DNA index range a separate cell by cell discriminate function analysis (DFA) was performed. The DFAs so generated were used to differentiate cells in normal samples from cells from abnormal samples in that DNA index range (see supplemental material, Appendix A). These DNA index boxes and DFAs were applied to all samples, and a calculation was made of the frequencies of cells in each category defined by the combination of DNA quantity and DFA. In this fashion 17 cell categories were defined (see Fig. 1). Fig. 1Definition of 17 cell categories: (a) Flow diagram of nuclei classification by DNA index thresholds and discriminate function analysis (DFA); and (b) application of this process to the nuclei in a CIS sample. In (a) all cells get divided into one of seven sets depending upon the amount of DNA detected in each cell (seven arrows leading away from all cells at top of figure). The cells in each of these sets gets further subdivided into two to three groups by a linear discriminate function unique to that set. See supplemental material Appendix A for a detailed description of the cell classification process. In (b) this process is applied to all the cells in a CIS sample, i.e. the cell classification process of initially separating cells into seven sets based upon their DNA amount, followed by the application of a DF specific to the individual DNA amount sets is applied so that each cell is assigned a DF score. The scatter plot of the cells in the CIS sample as a function of DNA amount and DF score is shown. 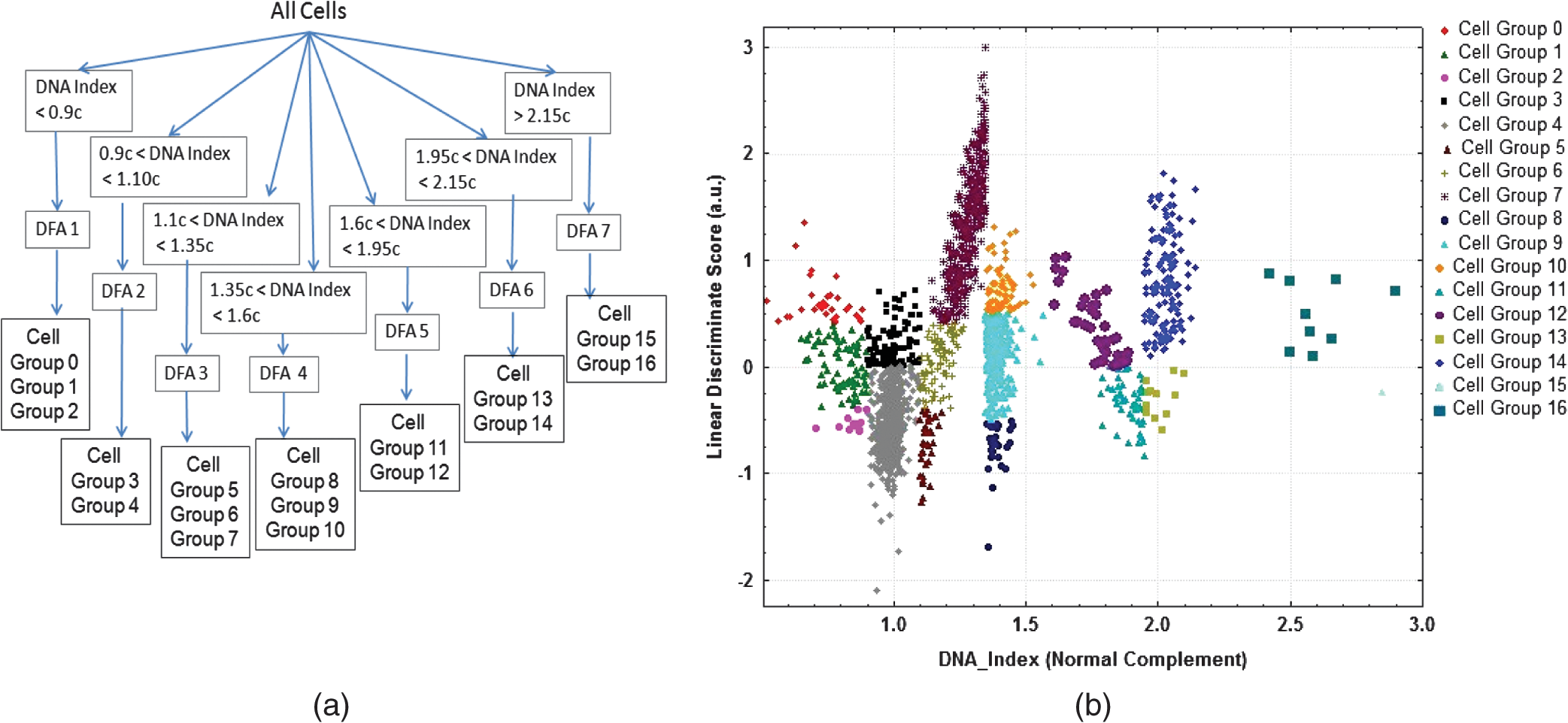 2.5.StatisticsThe overall approach used to differentiate normal samples from abnormal samples was to use the normal set to define ranges that represent normal limits; samples with characteristics outside these values were classified as “not” normal samples. The thresholds on the frequency of cells in these categories were used to separate normal samples from abnormal samples in the training set. values for the separation between the categories in the training sets were determined for all cell categories and scatter plots were made of the cell category frequencies that had the largest separation. The appropriate cutoffs for sample classification were heuristically evaluated from these graphs. Once these differentiating thresholds were set, the detection algorithms were run on all the samples. All statistical calculations, nonparametric Mann-Whitney U tests and Komogorov-Smirnov tests and scatter plots were produced using Statistica version 8.0 (StatSoft Inc., Tulsa, Oklahoma). A value of less than 0.05 was considered to be statistically significant and a value of less than 0.005 to be highly significant. 3.ResultsFigure 2 shows conventional white light and direct FV images of the oral cavity of a normal subject, a patient with a confounding lesion, and a patient with a CIS. In each case, a display of the most abnormal cell images (as called by the algorithm) collected from the brushing sample is displayed with a histogram of DNA content for the cells along with the number of cells classified into the 17 different cell categories. In Fig. 2(a) to 2(c), the oral tissue looks healthy under white light and has substantial green autofluorescence in the direct FV image. The corresponding cell images and narrow histogram show normal cells of which all are in the G0/G1 state (peak at DNA content of 1) and are classified into the cell categories associated with normal samples. In Fig. 2(d) to 2(f), a small white lesion is apparent under white light with a substantial loss of green autofluorescence immediately surrounding the lesion in the direct FV image. This is an example of a confounding lesion. The corresponding cell images and wider histogram (larger variance in DNA content) show normal cells of which most are in the G0/G1 state and are also classified into the cell categories associated with normal samples but with some indication of a small cycling cell population (cells with DNA index of 1.4 to 2). These changes are all consistent with a response to wounding or infection. In Fig. 2(g) to 2(i), we can see a lesion under white light with substantial loss of green autofluorescence in the direct FV image. The corresponding cell images and DNA content histogram indicate many cells that are not part of the peak at DNA content 1 and with classifications across all the cell categories. Many of the cells have large nuclei with DNA that is very irregularly distributed and disorganized indicating a substantial loss of higher order DNA and chromosomal organization. These changes are all consistent with the significant genetic alterations associated with oral cancer. Fig. 2Images (a), (d), and (g) show direct fluorescence visualization (FV) images of normal, inflammation and cancer, respectively. Images (b), (e), and (h) show the corresponding white light images of the same areas. The direct fluorescence visualization image of normal tissue has substantial green autofluorescence (a), the small white lesion under white light in (e) has substantial loss of green autofluorescence immediately surrounding the lesion (d) and similarly the lesion in (h) has a substantial loss of green autofluorescence immediately surrounding the lesion (g). Images (c), (f), and (i) display images of nuclei from each of the areas/lesions sampled along with histograms of DNA content. Image (c) shows a narrow histogram of cells in the G0/G1 state (peak at DNA content of 1), and most of the cells are classified into the cell category associated with normal samples (group 4, black arrow). Image (f) shows a wider histogram (larger variance in DNA content) but most cells are still in the G0/G1 state (groups 3 and 4, blue arrows) and are also classified into the cell categories associated with normal samples, but with some indication of a small cycling cell population (cells with DNA content of 2, group 14 and group 9, thin blue arrows). These changes are all consistent with a response to wounding or infection. In (i) the DNA content histogram indicates there are many cells which are not part of the peak at DNA content 1 and are also classified across all the cell categories (I, groups 7 to 16, red arrow). All the cell images in (i) show abnormal cells with approximately twice as much DNA as the normal complement, large in size, a few multinucleated cells and irregular texture.  4.Creation of Detection AlgorithmsA set of discriminate functions (DF) was developed to differentiate cells in the normal samples from cells from the abnormal samples in the training set. Cells were separated into the seven specific DNA content ranges as defined in Sec. 2. The DF training process was allowed to select up to seven features from within the 110 features available using a forward stepping feature selection process as part of the discriminate analysis, resulting in the creation of a DF for each of the DNA ranges. Thresholds were applied to the DF scores to subdefine cell categories within each DNA index box based upon the DF score as shown in Fig. 1. A visual display of the 17 cell categorizing classifications across a scatter plot of DNA index versus discriminate function score is shown in Fig. 1. All 17 cell categories showed a significant difference between normal and abnormal samples, with 15 of the 17 cell categories having values less than 0.0001 (see Table 1). Table 1Difference of frequency of cells in 17 categories between normal and abnormal samples in training set cases.
The frequency of cells classified into the 17 cell categories for the normal and abnormal cases was further examined, and cell categories were identified in which the frequency of cells were sufficiently different that a simple threshold could correctly classify at least 10% of one category while misclassifying none of the other categories. Cell categories 12 and 16 were found to classify the most abnormal cases while misclassifying none of the normal samples. Figure 3 is a scatter plot of the frequency of cells in each sample of the training set for these two categories. This graph was used to determine thresholds that separated normal samples from abnormal samples. In other words, if the frequency of cells in either category 12 or category 16 exceeded that cell category’s threshold, the sample would be classified as abnormal. Fig. 3A scatter plot of the frequency that cells occur in cell groups 12 and 16 for the samples from normal and abnormal (severe dysplasia, CIS, and SCC) sites in the training set. These are the two best cell groups found to differentiate between normal from abnormal samples. The inset graph is a magnified view of the lower left corner of the main graph. Note how all the normal samples are in the lower left corner of the graphs.  Previous studies have shown that a large number of cells were needed to define a sample as negative; however, even a few abnormal cells (“alarm” cells) are sufficient to recognize an abnormal sample. Automated analysis of cells from the cervix and sputum (previously reported) has found that 500 to 1000 cells per case were required for robust results.20,21 From an analysis of classification results using cell categories 12 and 16 (Fig. 3), a cut-off of at least 400 cells per case yielded good performance while minimizing the exclusion of samples due to inadequate cell numbers. For a sample to be classified as normal, it could not have cell frequencies exceeding the thresholds set for cell categories 12 and 16, and it had to have more than 400 cells measured. Otherwise the sample would be classified as inadequate. Sample adequacy across the different algorithms described below varied from 97.3% to 99%. Initially the confounders were part of the test set. Six different algorithms of increasing complexity (increasing numbers of cell categories used for case classification) were created. While all six of these algorithms generalized well in that the normal and abnormal test sets demonstrated the same sensitivity and specificity results as achieved in the training sets, their performance on the confounders decreased with increasing algorithm complexity. For the normal and abnormal sets, the sensitivity of the six algorithms ranged from 61% to 89% with specificity from 90% to 100%; at the same time, the specificity on the confounder set dropped from 89% to 48% (for detailed results please see supplemental material, Appendix B). Initially, we assumed that the variability within the normal training set would be representative of that present in the normal test set samples and confounder samples. These results suggested that this is not a valid assumption for the confounder set possibly because the biology that causes the tissue to be detected as a confounder alters the fraction of cycling cells within the tissue. This would have the effect of increasing the DNA variability observed within the cells from these samples. It is likely that trauma/infection/inflammation cause some fraction of the cells in the tissue to proliferate in response to the environmental assault. The data show that the confounder set has increased variability over the normal set and that thresholds designed to define the limits of normal incorrectly recognize some of the confounders as outside normal limits. To adjust for the poor performance on the confounders using the above approach, the threshold selection analysis was repeated, but the confounders were included as part of the normal training set when setting the category thresholds. Table 2 summarizes the results of this modification to the training. It also subdivides the results across the different types of confounders: trauma (23 sites), inflammation (49 sites), and infection (5 sites). The inclusion of the confounders in the training set improved the results for the confounders while maintaining the accuracy on the normal and abnormal training and test sets. Algorithm 9 correctly classifies the most samples in the test set at the same time yielding very good performance on the confounders. Table 2Classification performance for Algorithms 7 to 9.
Note: Algorithm 7 uses thresholds on the frequency of cells in cell groups 0+1+2, 3+4, 8+9+10, 11+12, 13+14, and 15+16 (uses DNA Index information only). Algorithm 8 uses slightly different thresholds on the frequency of cells in cell groups 0+1+2, 3+4, 8+9+10, 11+12, 13+14, and 15+16 (thus algorithm 8 only uses DNA Index information). Algorithm 9 uses thresholds on the frequency of cells in cell groups 3+4, 0 to 4, 6 and 9 to 16. Percentages indicate the frequency of samples correctly classified. Values in brackets are the number of samples that were found to be inadequate for analysis. Since Algorithm 9 was intended to be used in a future prospective study, its performance was examined with respect to patient demographics including gender and smoking status. An analysis of the data shown in Table 3 resulted in no statistically significant association with these demographic variables. Table 3Algorithm 9 classification performance stratified by gender and smoking status.
5.Use of Algorithms for FV Image InterpretationOne of the possible uses of quantitative cytology (QC) is to facilitate the correct identification of confounders encountered with FV. FV data was available for only 307 of 369 (83%) of the samples in this study including 140 of 158 (89%) of the abnormal samples, 65 of 77 (84%) of the confounder samples, and 158 of 161 (98%) of the normal samples. Data were limited for some categories. Only two abnormals were FV-negative (no loss of autofluorescence apparent), only one normal was FV positive (loss of autofluorescence apparent), eight confounders were FV-negative and 57 confounders were FV positive. Of the 57 FV positive confounders, only seven were also positive using Algorithm 9. Thus QC recognized 88% of the confounders correctly. There is a positive correlation between QC and FV across the normal and confounders sets (). This suggests that perhaps inflammation/infection/trauma detected by FV correlates with slightly increased cell proliferation. Further there was no statistical difference () between the performance of QC within the FV positive and FV negative categories with respect to the detection of positive at-risk tissue. There was no correlation within the abnormal training and test sets () with respect to QC and FV. Thus QC and FV appear to behave as independent detector methodologies. 6.DiscussionIn British Columbia, the Oral Cancer Prevention Program has evaluated several oral cancer screening technologies with the intent of implementing a comprehensive screening system that identifies patients within community dental practices for referral to cancer care centers. Heterogeneity in the clinical presentation of oral lesions can make their assessment challenging even for experienced oral specialists. Such ambiguous lesions are more frequent in a community setting than true at-risk tissue and as there is some morbidity associated with tissue removal from the oral cavity, clinicians can be hesitant to biopsy. As part of the evolution of new visualization technology, the focus has been on ambiguous lesions more likely to be seen in the community setting with conventional examination or by direct fluorescence visualization. This paper describes the development of a minimally invasive brushing based tool for detection of lesions that require subsequent follow-up. The data presented in this paper demonstrate how the combination of DNA quantity and organization/texture features can be used to facilitate the differentiation of lesions with severe dysplasia and higher pathology from infection and inflammation without requiring the presence of frankly abnormal cells, which can be rare even in samples collected directly from the most altered area of the lesion and are even less abundant in less selectively collected samples. The approach taken in this paper was designed from the beginning to be robust to a less than perfectly targeted sample acquisition, i.e., enabling it to be able to recognize cells coming from patients with at-risk tissue as opposed to cells coming from sites of infection, inflammation, and trauma even if the brushing was not taken directly from the target tissue but from the immediate surrounding tissue. 6.1.Comparison with Other Oral Cytology Assessment ApproachesGenetic instability is a central hallmark of cancer.22–25 Mutations, LOH, and aneuploidy can all play a role in the uncontrolled growth of cancer. Detecting these changes to the genome is one important way to follow the progression of cancer and to catch clinical cancer (cancer and its precursor lesions, which are at sufficient risk of becoming malignant that they are treated clinically, usually surgically) early. Aneuploidy occurs when a cell has an abnormal chromosomal content or number. This can arise from gain or loss of whole chromosomes or by gross alteration of one or more chromosomes through deletions, translocations, end to end fusions, or other events.26,27 Polysomes of chromosomes 7, 9, and 17 increase as oral epithelial tissue progresses from low risk lesions (hyperplasia) to high risk (severe dysplasia).28,29 There is a long history for the use of DNA amount measurements as detectors of malignancy. Several research groups have attempted to quantify the large scale genetic instability (aneuploidy) that develops with oral tumorgenesis as a means to assess cancer risk. Using cells dissociated from formalin-fixed paraffin-embedded tissue from 45 primary oral cancer, Diwakar et al. looked at not only the ploidy status of the oral tumors, but the consistency of the ploidy classification across the tumor.30 Their work demonstrated that a definition of nondiploid would correctly identify 93.3% (42 out of the 45) of oral cancers. Researchers from Dűsseldorf have combined clinical cytological examination and DNA image cytometry of disaggregated tissue to differentiate high risk oral lesions (severe dysplasia, CIS, cancer) from normal tissue in one study31 and in a separate study to assess the risk of cancer in confounding lichen planus lesions.32 Using this combined cytological/cytometric method, they reported a very encouraging sensitivity of 100% and a specificity of 97.4% for high risk lesions that were either DNA-aneuploidy, or cytologically suspicious.31 Furthermore, only 2 of 56 lichen planus were positive, and these were shown to have an SCC component in complete agreement with their histological findings. While this approach showed high sensitivity and specificity, the heavily manual aspects (pathology expertise) limits the high throughput needed to be clinically relevant. In both of these studies, oral epithelial cells were obtained by targeted brushing, then stained, and graded by a pathologist. The DNA level was then quantified on those samples deemed visually suspect using computer-based image cytometry. In addition when Pektas et al. tried to duplicate this approach, using a manual cytometric DNA image analysis system coupled to expert clinical cytological assessment, only 16.7% of malignant oral lesion samples collected by cytobrushing were classified as aneuploid,33 indicating that the sensitivity of this form of combined analysis can be quite variable. In contrast, our high throughput automated image processing system requires very little human interaction, making it possible for a single operator to assess many samples daily while maintaining a performance similar to that reported by other groups that required input from skilled clinical specialists to perform the analysis. 6.2.Use of DNA Organization to Assist in Sample ClassificationTo maximize the diagnostic information available beyond that available in the DNA index from the cytological samples, algorithms were developed that differentiated between normal cycling cells and cells altered as part of the neoplastic process as described previously. This approach, which is based upon the direct detection of genetic and epigenetic alterations in the DNA within nuclei, has been accomplished through the use of measures of nuclear DNA organization as recorded through image texture features. Specifically, these algorithms attempted to glean such further discriminating ability from the measured cells by calculating linear discriminate functions (LDF) for each DNA range to differentiate between cycling and aneuploid cells. Similar to most epithelial tissue, the oral cavity has a high enough tissue renewal rate that the presence of cells in non G0/G1 states is not a rare event. As part of the neoplastic development process, cells can acquire inheritable alterations, which alter their DNA content such that it is discernibly more than that of G2/M cells, making them easily identified as abnormal cells. It is the altered cells without such frank DNA changes that are challenging to recognize. If one can differentiate between cycling cells and true aneuploid (abnormal) cells increased sample classification is possible. Our LDFs made use of five to seven features selected from the over 100 available; generally the selected features contained one or two nuclear shape features with the rest being texture features that quantify the appearance of the DNA within the nuclei. Specifically these features describe DNA organization deregulation (hyperchromasia) associated with molecular and genetic alterations involved with the neoplastic process at the cellular level. This is the first time that nuclear hyperchromatism as quantified by DNA texture measures has been integrated into the interpretation of oral cytology. Others have shown that increased proportion of heterochromatin condensation is a high-risk nuclear characteristic for cancer.28 Our previous quantitative histology work showed that the quantification of the many facets of heterochromatin condensation and hyperchromatism is predictive of cancer development and is strongly associated with genetic level alterations within oral tissue.5–7,34 Thus it was not unexpected that the training process for the LDFs resulted in the selection of features that are known to be measures of nuclear hyperchromasia. This approach can differentiate between oral brushing samples from normal and abnormal sites even when the sampling is not directly targeted to only the area that an experienced oral specialist would select. It is robust to even include cells from the immediate surrounding tissue, such as those a clinician who is infrequently exposed to oral lesions might select. For brushing cytology samples collected only from the area selected by an experienced oral specialist and hence not diluted by the surrounding normal cells one would reasonably expect the system to perform even better. The performance of the final algorithm while validated on the normal and abnormal test sets has not been validated on a confounder test set. This work needs to be reproduced in an independent test set. Such studies would be strengthened if they occurred in dental practices with dental health professionals as screeners. An efficient way to evaluate this next step would be to use QC in high-risk community settings, where cancer and dysplasia rates are known to be elevated, such as in communities characterized by high frequencies of habit usage and low socioeconomic status. Confounders are also high in such settings making cancer detection more difficult but making such a setting an excellent place for validation. An ongoing study is running in this setting to further evaluate the value of QC. Initial results on a subset of 20 cases of these confounding cases (confounder test set) achieved essentially the same results as shown in Table 2 for Algorithm 9. 6.3.Combining Macroscopic and Microscopic ImagingAn important aspect of this study is that it demonstrates that the layering of clinical evaluation of the oral cavity using wide-field white light and/or FV with image cytometry of approximately targeted brushings can differentiate reactive and inflammatory conditions from true at-risk tissue (Fig. 2). In a clinical practice setting one initially uses a wide-field examination (conventional white light and/or fluorescence visualization) followed by a point sampling methodology (currently biopsy). The technology introduced here is a bridge in that it is not an invasive biopsy procedure but does acquire some cellular material for interrogation. The morbidity associated with brushing is minimal. As such it will be better tolerated in the dental practice setting and is eminently amenable to practice as a high throughput low-cost platform. This is exactly how this same platform is being used for cervical screening in resource-limited settings such as China35 where it has been used to screen more than 400,000 women. A separate but related benefit associated with QC is that it is readily scalable so it can handle a potential increase in referral of confounding lesions such as might occur with the introduction of a new wide field screening methodology such as FV. A stratification of the results presented here by FV status showed no difference in the ability of the QC to detect at risk tissue appropriately in the absence or presence of fluorescence loss (). These data support the potential utility of the coupled use of FV and automated image cytometry for the detection of oral cancer. As such it is not intended to replace biopsy for the obvious neoplastic high-risk tissue. We envisage its primary usage in allowing dentists to query if the ambiguous lesion is at-risk. The data suggest that QC could reduce by more than 85% the number of lesions required to go forward to further assessment by biopsy. In summary, opportunistic screening that takes place within a routine dental check-up is widely considered the most cost-effective screening strategy. However, correctly identifying and classifying oral lesions, especially when inflamed, is difficult even for trained pathologists.36 Our data suggest that a high-throughput automated cytological image analysis system, based on targeted brushing of suspect lesions, could be an efficient and effective second step in a comprehensive screening program, directing the high risk patient populations to appropriate care while not necessitating a large number of biopsies. Such an approach could facilitate the widespread use of opportunistic screening to effectively manage oral cancer. AcknowledgmentsGrant support: The National Institute of Health and the National Institute of Dental and Craniofacial Research (R01DE13124 and R01DE17013). CFP is supported by Michael Smith Foundation for Health Research (Scholar Award). AppendicesAppendix A:Cell by Cell Classification By DNA Index Range and Discriminate FunctionsApproximately 10,000 cells at random from the over 175,000 cells making up all the cell data in only the normal training and abnormal training samples were selected to be used to train the following cell by cell DFA classifiers. These 10,000 cells were subdivided into seven sets with discrete DNA index ranges: less than 0.9 (set 1), 0.9 to 1.1 (set 2), 1.1 to 1.35 (set 3), 1.35 to 1.6 (set 4), 1.6 to 1.95 (set 5), 1.95 to 2.15 (set 6) and 2.15 and higher (set 7). Each of these sets contains cells from normal samples and cells from abnormal samples spanning the training sample set. For each of these seven cell sets a forward stepping feature selection as part of a linear discriminate function analysis was performed to differentiate the cells from normal samples from cells from abnormal samples. While the number of cells available is large, we limited the number of features allowed to be in the discriminate function by selecting large to enter and to remove values to reduce the possibility of over training. The value for a variable used in a step wise analysis indicates its statistical significance to discriminate between the groups; it is a measure of the extent a variable could make a unique contribution to the prediction of group membership. The actual features included in each discriminate function for the seven DNA ranges are listed below along with a brief description of the features used (see Refs. 18 and 37 for more complete feature descriptions). A1.Set 1 DFA Results (Applied to Cells with DNA Amount Less than 0.9)Eight cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Elongation (shape feature), Harmon06_fft (shape feature, size of 6th harmonic from a fast Fourier transform, fft, of nucleus radius as function of angle), DNA_Index (amount of DNA in cell), OD variance (variance of OD of pixels in nuclei), hiDNAamount (fraction of DNA in very condensed state in nucleus), Low av dst (average distance from nucleus center of noncondensed DNA), Correlation (cooccurrence matrix feature measuring intensity correlation of adjacent pixels in nucleus) and fractal dimension (measure of DNA texture). The range of the DF scores for the cells was divided into three parts so that the DF subdivided the cells in set 1 into 3 cell categories (0, 1, and 2). A2.Set 2 DFA Results (Applied to Cells with DNA Amount from 0.9 to 1.1)Seven cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: OD skewness (skewness of OD of pixels in nuclei), mhDNA amount (fraction of DNA in condensed state in nucleus), cl_shade (cooccurrence matrix feature measuring degree of correlation of adjacent pixels in nucleus), Den_drk_Spot (number of dark spots in nucleus/area of nucleus), Fractal1_area (see Ref. 18), Min_long _runs (see Ref. 18), and Min_gray_level (see Ref. 18). The range of the DF scores for the cells was divided into two parts such that the cells in set 2 were subdivided into two cell categories (3 and 4). A3.Set 3 DFA Results (Applied to Cells with DNA Amount from 1.1 to 1.35)Five cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Harmon03_fft (shape feature, size of 3rd harmonic from the fft of nucleus radius as function of angle), DNA_Index (amount of DNA in cell), OD maximum (maximum OD of pixels in nuclei), hiDNAarea (fraction of area of nucleus in which the DNA is in very condensed state), and Den_drk_Spot (number of dark spots in nucleus/area of nucleus). The range of the DF scores for the cells was divided into three parts so that the DF subdivided the cells in set 3 into three cell categories (5, 6, and 7). A4.Set 4 DFA Results (Applied to Cells with DNA Amount from 1.35 to 1.6)Five cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Freq_high_fft (shape feature-measure of small irregularities in nuclear border, sum of the amplitudes of the higher harmonics from fft of nucleus radius as function of angle), OD variance (variance of OD of pixels in nuclei), Contrast (cooccurrence matrix feature measuring intensity of contrast between adjacent pixels in nucleus), Fractal2_area (see Ref. 18) and Min_short_runs (see Ref. 18). The range of the DF scores for the cells was divided into three parts so that the DF subdivided the cells in set 4 into three cell categories (8, 9, and 10) A5.Set 5 DFA Results (Applied to Cells with DNA Amount from 1.6 to 1.95)Five cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Compactness (shape feature measuring closeness of nucleus shape to a circle), Harmon07_fft (shape feature, size of 7th harmonic from fft, of nucleus radius as function of angle), DNA_Index (amount of DNA in cell) cl_shade (cooccurrence matrix feature measuring degree of correlation of adjacent pixels in nucleus) and size_txt_orientation (degree of orientation polarization of the intensity changes in the nucleus). The range of the DF scores for the cells was divided into two parts such that the cells in set 5 were subdivided into two cell categories (11 and 12). A6.Set 6 DFA Results (Applied to Cells with DNA Amount from 1.95 to 2.15)Five cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Elongation (shape feature measuring ratio of major axis divided by the minor axis of an ellipse fit to the nucleus), Harmon03_fft (shape feature, size of 3rd harmonic from fft, of nucleus radius as function of angle), medDNAamount (fraction of DNA between a condensed and noncondensed state in nucleus), Correlation (cooccurrence matrix feature measuring intensity correlation of adjacent pixels in nucleus) and cl_prominence (cooccurrence matrix feature measuring degree of correlation of adjacent pixels in nucleus). The range of the DF scores for the cells was divided into two parts so that the DF scores could be used to subdivide set 6 into two cell categories (13 and 14). A7.Set 7 DFA Results (Applied to Cells with DNA Amount Greater than 2.15)Five cell features were combined in a linear discriminate function (DF) to score the cells into normal or abnormal categories: Mean_radius (shape feature measuring average radius of the nucleus), Harmon06_fft (shape feature, size of 6th harmonic from fft of nucleus radius as function of angle), lowDNAcomp (compactness, perimeter/area, of DNA in a noncondensed state in nucleus), lowVSmedDNA (ratio of DNA in noncondensed state versus DNA between a condensed and noncondensed state in nucleus) and center_of_grav (difference between geometric center based only on nuclear shape and center of mass of the DNA in the nucleus). The range of the DF scores for the cells was divided into two parts so that the cells in set 7 were subdivided into two cell categories (15 and 16). Once all these discriminate functions were trained on the 10,000 cells selected from the training cases they were applied to all cells in all samples classifying the cells from each case into one of the 17 cell categories. The frequency of cells in these categories was then used to classify the individual samples. AppendicesAppendix B:Case by Case Classification AlgorithmsThe results for the first six algorithms are shown in Table 4. Algorithm 1 uses thresholds on the frequency of cells in cell groups 12 and 16. Algorithm 2 uses thresholds on the frequency of cells in cell groups 12, 13, and 16. Algorithm 3 uses thresholds on the frequency of cells in cell groups and (thus uses DNA Index information only). Algorithm 4 uses thresholds on the frequency of cells in cell groups 3, 4, and 8 to 16. Algorithm 5 uses thresholds on the frequency of cells in cell groups , , , and (uses DNA Index information only). Algorithm 6 uses thresholds on the frequency of cells in cell groups 0, 3, 4, and 8 to 16. Percentages indicate the frequency of samples correctly classified. Values in brackets are the number of samples that were inadequate for analysis. Table 4Classification of Samples Algorithms 1 to 6.
ReferencesP. Laneet al.,
“Simple device for the direct visualization of oral-cavity tissue fluorescence,”
J. Biomed. Opt., 11
(2), 024006
(2006). http://dx.doi.org/10.1117/1.2193157 JBOPFO 1083-3668 Google Scholar
D. Roblyeret al.,
“Objective detection and delineation of oral neoplasia using autofluorescence imaging,”
Cancer Prev. Res., 2
(5), 423
–431
(2009). http://dx.doi.org/10.1158/1940-6207.CAPR-08-0229 CPRACC 1940-6207 Google Scholar
A. Milbourneet al.,
“Results of a pilot study of multispectral digital colposcopy for the in vivo detection of cervical intraepithelial neoplasia,”
Gynecol. Oncol., 99
(3 Suppl. 1), S67
–S75
(2005). http://dx.doi.org/10.1016/j.ygyno.2005.07.047 GYNOA3 Google Scholar
D. Pellman,
“Aneuploidy and cancer,”
Nature, 446
(7131), 38
–39
(2007). http://dx.doi.org/10.1038/446038a NATUAS 0028-0836 Google Scholar
M. Guillaudet al.,
“Potential use of quantitative tissue phenotype to predict alignant risk for oral premalignant lesions,”
Cancer Res., 68
(9), 3099
–3107
(2008). http://dx.doi.org/10.1158/0008-5472.CAN-07-2113 CNREA8 0008-5472 Google Scholar
M. Guillaudet al.,
“Nuclear morphometry as a biomarker for bronchial intraepithelial neoplasia: correlation with genetic damage and cancer development,”
Cytometry A, 63
(1), 34
–40
(2005). http://dx.doi.org/10.1002/(ISSN)1552-4930 1552-4922 Google Scholar
C. Thibervilleet al.,
“Multi-scale system biology applied to cervical inter-epithelial neoplasia,”
Gynecol. Oncol., 107
(1 Suppl. 1), S72
–S82
(2007). http://dx.doi.org/10.1016/j.ygyno.2007.07.047 GYNOA3 Google Scholar
M. Rosinet al.,
“New hope for an oral cancer solution: together we can make a difference,”
J. Can. Dent. Assoc., 74
(3), 261
–266
(2008). JCDAAS 0008-3372 Google Scholar
D. M. Laronde,
“Improving the detection and triage of oral premalignant lesions in high-risk clinics and community dental practices,”
Simon Fraser University,
(2009). Google Scholar
S. L. WilliamsonT. HairV. Wadehra,
“The effects of different sampling techniques on smear quality and the diagnosis of cytological abnormalities in cervical screening,”
Cytopathology, 8
(3), 188
–195
(1997). http://dx.doi.org/10.1046/j.1365-2303.1997.4675046.x CYTPEU 1365-2303 Google Scholar
P. Martin-Hirschet al.,
“Collection devices for obtaining cervical cytology samples,”
Cochrane Database Syst. Rev., 2000
(3), CD001036
(2007). Google Scholar
B. Palcicet al.,
“Oncometrics Imaging Corp. and Xillix Technologies Corp.: use of the Cyto-Savant in quantitative cytology,”
Acta Cytologica, 40
(1), 67
–72
(1996). http://dx.doi.org/10.1159/000333599 ACYTAN 0001-5547 Google Scholar
D. M. Garneret al.,
“Cyto-Savant™ and its use in automated screening of cervical smears,”
Compendium on the Computerized Cytology and Histology Laboratory, 346
–352 Tutorials of Cytology, Chicago
(1994). Google Scholar
F. Giroudet al.,
“Part II.Specific recommendations for quality assurance. The 1997 ESCAP consensus report on diagnostic DNA image cytometry,”
Anal. Cell. Pathol., 17
(4), 201
–208
(1998). ACPAER 0921-8912 Google Scholar
D. Chiuet al.,
“Quality assurance using statistical process control: an implementation for image cytometry,”
Cellular Oncol., 26
(3), 101
–117
(2004). COENGH 2211-3436 Google Scholar
G. Andersonet al.,
“The use of an automated image cytometer for screening and quantitative assessment of cervical lesions in the B.C. cervical smear screening programme,”
Cytopath., 8
(5), 298
–312
(1997). http://dx.doi.org/10.1111/cyt.1997.8.issue-5 CYTPEU 1365-2303 Google Scholar
A. Doudkineet al.,
“Nuclear texture measurements in image cytometry,”
Pathologica, 87
(3), 286
–289
(1995). PATHAB 0031-2983 Google Scholar
G. Bradleyet al.,
“Abnormal DNA content in oral epithelial dysplasia is associated with increased risk of progression to carcinoma,”
Br. J. Cancer, 103
(9), 1432
–1442
(2010). http://dx.doi.org/10.1038/sj.bjc.6605905 BJCAAI 0007-0920 Google Scholar
G. Liet al.,
“Automated sputum cytometry for detection of intraepithelial neoplasias in the lung,”
Analyt. Cellular Pathol., 35
(3), 187
–201
(2012). ACPAER 0921-8912 Google Scholar
M. Guillaudet al.,
“DNA ploidy compared to human papilloma virus testing (Hybrid Cature II) and conventional cervical cytology as a primary screening test for cervical high grade lesions and cancer in 1555 patients with biopsy confirmation,”
Cancer, 107
(2), 309
–318
(2006). http://dx.doi.org/10.1002/(ISSN)1097-0142 60IXAH 0008-543X Google Scholar
M. E. Pretoriuset al.,
“Large scale genomic instability as an additive prognostic marker in early prostate cancer,”
Cellular Oncol., 31
(4), 251
–259
(2009). http://dx.doi.org/10.3233/CLO-2009-0463 COENGH 2211-3436 Google Scholar
A. Fabariuset al.,
“Genomic instability in context of the chromosomal theory, Letter to the Editor,”
Cellular Oncol., 30
(6), 503
–504
(2008). COENGH 2211-3436 Google Scholar
H. Q. Henget al.,
“Patterns of genomic dynamics and cancer evolution, Letter to the Editor,”
Cellular Oncol., 30
(6), 513
–514
(2008). COENGH 2211-3436 Google Scholar
M. R. Teixeira,
“Multiple numerical chromosome aberrations in carcinogenesis: the kidney cancer model, Letter to the Editor,”
Cellular Oncol., 31
(1), 57
–59
(2009). COENGH 2211-3436 Google Scholar
G. J. P. L. KopsB. A. A. WeaverD. W. Cleveland,
“On the road to cancer: Aneuploidy and the mitotic checkpoint,”
Nat. Rev. Cancer, 5
(10), 773
–85
(2005). http://dx.doi.org/10.1038/nrc1714 NRCAC4 1474-175X Google Scholar
W. N. Hillelman,
“Genetic instability in epithelial tissues at risk for cancer,”
Ann. N. Y. Acad. Sci., 952 1
–12
(2001). http://dx.doi.org/10.1111/j.1749-6632.2001.tb02723.x ANYAA9 0077-8923 Google Scholar
N. Voravudet al.,
“Increased polysomies of chromosomes 7 and 17 during head and neck multistage tumorigenesis,”
Cancer Res., 53
(12), 2874
–2883
(1993). CNREA8 0008-5472 Google Scholar
J. Kimet al.,
“Chromosome polysomy and histological characteristics in oral premalignant lesions,”
Cancer Epidemiol. Biomarkers Prev., 10
(4), 319
–25
(2001). CEBPE4 1055-9965 Google Scholar
N. Diwakaret al.,
“Heterogeneity, histological features and DNA ploidy in oral carcinoma by image-based analysis,”
Oral Oncol., 41
(4), 416
–22
(2005). http://dx.doi.org/10.1016/j.oraloncology.2004.10.009 EJCCER 1368-8375 Google Scholar
D. MarakiJ. BeckerA. Boecking,
“Cytologic and DNA-cytometric very early diagnosis of oral cancer,”
J. Oral Pathol. Med., 33
(7), 398
–404
(2004). http://dx.doi.org/10.1111/j.1600-0714.2004.0235.x JPMEEA 0904-2512 Google Scholar
D. Marakiet al.,
“Cytologic and DNA-cytometric examination of oral lesions in lichen planus,”
J. Oral Pathol. Med., 35
(4), 227
–32
(2006). http://dx.doi.org/10.1111/jop.2006.35.issue-4 JPMEEA 0904-2512 Google Scholar
Z. Ö. Pektaset al.,
“Evaluation of nuclear morphometry and DNA ploidy status for detection of malignant and premalignant oral lesions: quantitative cytologic assessment and review of methods for cytomorphometric measurements,”
J. Oral. Maxillofax. Surg., 64
(4), 628
–35
(2006). http://dx.doi.org/10.1016/j.joms.2005.12.010 JOMSDA 0278-2391 Google Scholar
C. MacAulayet al.,
“Malignancy-associated changes in bronchial epithelial cells in biopsy specimens,”
Anal. Quant. Pathol. Histol., 17
(1), 55
–61
(1995). Google Scholar
X. R. Sunet al.,
“Detection of cervical cancer and high grade neoplastic lesions by a combination of liquid-based sampling preparation and DNA measurements using automated image cytometry,”
Cell Oncol., 27
(1), 33
–41
(2005). COENGH 2211-3436 Google Scholar
D. J. Fischeret al.,
“Interobserver reliability in the histopathologic diagnosis of oral pre-malignant and malignant lesions,”
J. Oral. Pathol. Med., 33
(2), 65
–70
(2004). http://dx.doi.org/10.1111/j.1600-0714.2004.0037n.x JPMEEA 0904-2512 Google Scholar
C. MacAulay,
“Development, implementation and evaluation of segmentation algorithms for the automatic classification of cervical cells,”
University of British Columbia,
(1989). Google Scholar
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



