
|
|
1.IntroductionOptical imaging is a powerful tool for monitoring the metabolic status of cells and tissues due to the auto-fluorescence properties of the metabolic co-enzymes NADH and FAD. NADH is the principal electron acceptor in glycolysis and electron donor in oxidative phosphorylation, while FAD is the primary electron acceptor in oxidative phosphorylation. The optical “redox ratio” (fluorescence intensity of NADH divided by that of FAD) is widely used to monitor cellular metabolism in cells, ex vivo tissues, and in vivo tissues in order to diagnose disease,1,2 monitor cellular differentiation,3 and characterize other metabolic perturbations.4,5 Recently, the fluorescence lifetimes of NADH and FAD have also been evaluated as optical metabolic biomarkers.2,6–10 The fluorescence lifetime is the time a fluorophore remains in the excited state before returning to the ground state and emitting a fluorescent photon. Fluorescence lifetimes are self-referenced and more robust than fluorescence intensity measurements in regard to inter-system and intra-system variability.7 Additionally, fluorescence lifetimes are sensitive to changes in pH, temperature, and proximity to quenchers.7,11 Often, the fluorescence lifetime decay curves for NADH and FAD are each fit to a two-component exponential decay.2,6,12 The two lifetime components represent the two most probable configurations of NADH and FAD, which are free in solution or bound to a protein.12,13 The short lifetimes and long lifetimes of NADH represent the free and protein-bound components, respectively. Conversely, the short and long lifetimes of FAD represent the protein-bound and free components, respectively. For these reasons, the fluorescence lifetimes of NADH and FAD are robust reporters for metabolic differences between tissues. Indeed, auto-fluorescence lifetime imaging has been used for monitoring pre-cancers and cancers in oral cancer models and in breast cancer models.2,6,10,14–18 Ideally, optical metabolic measurements should be performed in vivo. Thus, these techniques have been predominantly used in easily accessible tissues, such as the skin and oral cancer models.2,8,19,20 The growing development of endoscopes capable of confocal fluorescence microscopy is enabling measurements of previously inaccessible tissue.21,22 However, fluorescence endoscopes are still somewhat specialized, cumbersome, and represent a significant technical and regulatory burden for feasibility studies, especially in tissue sites that are difficult to access (i.e., outside of the gastrointestinal tract). As a result, many feasibility studies in this field are performed on fixed tissue slides, immortalized cell lines, or excised tissue,6,15,23 which may not accurately represent the metabolic state of live, intact tissue. Furthermore, the use of optical metabolic techniques would be greatly expanded by the development of an effective tissue handling protocol that allowed for ex vivo tissue interrogation that accurately reflects the in vivo metabolic state. However, the effect of ex vivo sample preparation on optical metabolic measurements has not been explored. The goal of this study was to investigate live tissue culture as an alternative sample preparation for optical metabolic imaging when in vivo measurements are not feasible. Current protocols to represent in vivo metabolic activity in excised tissue require flash-freezing of the tissue;4 however, such a procedure requires tissue submersion in liquid nitrogen and may induce changes in sensitive fluorescence lifetime endpoints. Live tissue culture may address these limitations. Live tissue culture maintains the tissue in tissue culture media that provides oxygen, glucose, and other necessary nutrients. Furthermore, chilled tissue media has been used in previous optical studies23,24 because it contains more dissolved oxygen than media maintained at body temperature. However, this chilled media procedure for maintaining tissue culture has not been rigorously compared to in vivo metabolic measurements. We hypothesized that, for a discrete period of time following harvest, excised tissue maintained in chilled tissue culture media will provide metabolic measurements that are representative of the in vivo metabolic state. To test this hypothesis, the NADH and FAD fluorescence intensity and lifetime, as well as cellular morphology, of the cheek pouch epithelium of 16 Golden Syrian hamsters were measured in vivo, ex vivo from biopsies maintained in live culture over 48 h, and ex vivo from flash-frozen and thawed tissue biopsies. Both high and low time-resolution protocols were followed to investigate both short-term and long-term changes in the cellular metabolism of the cultured biopsies. For the high-resolution protocol, optical metabolic measurements were obtained every 30 min for 4 h from a single epithelial depth. For the low-resolution protocol, images of two different epithelial depths were acquired at 4, 8, 12, 24, and 48 h. 2.Materials and Methods2.1.Hamster ProtocolFor this study, 16 male Golden Syrian hamsters were evaluated (). This study was approved by the Vanderbilt University Animal Care and Use Committee and meets the National Institutes of Health guidelines for animal welfare. Hamsters were anesthetized with a mixture of ketamine and xylazine with an intraperitoneal injection. The mucosa of the cheek pouch was exposed and secured against a coverslip for imaging. All images were acquired from the stratum spinosum or basal epithelial layers at depths of approximately 20 or 30 μm, respectively. In vivo measurements of the cheek pouch were obtained from the cheek epithelium. Then, two 6-mm biopsies of the cheek pouch were acquired immediately after the in vivo measurements from the same region that was imaged in vivo. For 10 hamsters, one biopsy per hamster was flash-frozen in liquid nitrogen (frozen-thawed sample). This sample was placed immediately in a freezer. The second 6-mm biopsy (live tissue culture sample) was immediately placed in a glass-bottom petri dish with chilled (3°C), sterile tissue culture media [Dulbecco’s Modified Eagle Medium (DMEM) without phenol red; GIBCO, Grand Island, New York). The stratum spinosum layer of the epithelium was immediately measured and sequentially measured every 30 min for 4 h after the time of the biopsy from the live tissue culture sample. For this high time-resolution protocol, a single tissue layer was interrogated to allow for comparisons between time points and to minimize tissue warming at room temperature during imaging. This high time-resolution, short time-frame protocol was chosen to investigate acute changes in the metabolism of the tissue immediately after excision. For the remaining six hamsters, after in vivo imaging, two biopsies were obtained and both were placed in chilled tissue media for the long-term (low time-resolution) imaging studies. This low time-resolution, long time-frame protocol investigates the maximum duration over which optical metabolic measurements in live culture correspond with in vivo measurements. One of the biopsies was imaged after 4, 8, 12, 24, and 48 h at two different depths corresponding to the spinous and basal regions. The second biopsy was fixed in formalin for histological analysis after 4, 8, 12, 24, or 48 h in live culture. Each measurement in live tissue culture over this time course was conducted within 5 min at room temperature (22°C), and the sample was refrigerated (3°C) between measurements. Prior to measurement of the frozen biopsy, the specimen was thawed in chilled tissue culture media for 15 to 20 min.25 Each animal was sacrificed after both biopsies had been obtained. 2.2.Imaging InstrumentationAll data were acquired using a custom-built, commercial two-photon imaging system (Prairie Technologies, Middleton, Wisconsin) within the Biophotonics Laboratory at Vanderbilt University. A titanium sapphire laser (Coherent Inc., Santa Clara, California) provided the excitation source. The excitation and emission light were coupled through an inverted microscope (TiE; Nikon, Melville, New York) with either a water-immersion objective [1.15 numerical aperture (NA)] or a oil-immersion objective (1.3 NA). For NADH excitation and FAD excitation, the laser was tuned to 750 and 890 nm, respectively. The average power incident on the sample was to 7.8 mW for NADH excitation and to 8.6 mW for FAD excitation. A pixel dwell time of 4.8 μs was used for all images. NADH and FAD images were acquired sequentially. A 400- to 480-nm bandpass filter isolated NADH emission onto the photomultiplier tube (PMT) detector. A dichroic mirror directed wavelengths greater than 500 nm onto a 500- to 600-nm bandpass filter, thus removing collagen second harmonic generation (SHG) signals from the FAD image. Fluorescence lifetime images were acquired with time-correlated single photon counting electronics (SPC-150; Becker and Hickl, Germany) and a GaAsP PMT (Hamamatsu, Japan). Isolation of NADH and FAD collection for this system was confirmed by treating cells with cyanide26 and observing an increase in NADH fluorescence intensity and a decrease in FAD fluorescence intensity (data not shown). The instrument response function was measured by imaging the SHG signal of urea crystals with excitation at 900 nm. The instrument system response has a full width at half-maximum of 260 ps. This measured system response was used in the lifetime fit model. The fluorescence lifetime of a standard Fluoresbrite YG microsphere (20 μm; Polysciences Inc.) was measured daily. Fit to a single decay curve, the fluorescence lifetime of the microsphere has a value of (), which is consistent with previous studies.2,6,10,27 2.3.Fluorescence Lifetime ImagingFor the in vivo measurement, the hamster was placed face down with the cheek pouch mucosa in contact with a coverslip and exposed to the objective. The epithelium of the cheek pouch was identified and imaged at a depth of . This depth was imaged across all samples (in vivo, ex vivo, and frozen tissues), as determined by stage translation distance from the surface of the tissue and by visual inspection of cell size. The six hamsters imaged for the longer live culture time course were also interrogated at a depth of , corresponding to the basal epithelium. One NADH lifetime measurement was acquired over an integration time of 60 s. Sequentially, an FAD lifetime image was acquired, also for 60 s, at the same location. Imaging parameters such as laser power, PMT voltage, pixel integration time, and lifetime collection time were maintained across all animals and measurements including the ex vivo and frozen samples. Photon count rates () were maintained throughout all imaging sessions, ensuring that photobleaching did not occur. In addition, it was assured that no quantifiable change in fluorescence intensity was observed between sequential images of the same location, further confirming that photobleaching did not occur. 2.4.Quantification of Cellular MorphologyIn order to monitor changes in cellular morphology in the live tissue culture, cell size and nuclear-cytoplasmic ratio (NCR) were computed from the high-resolution NADH fluorescence images. A region of well-defined cells was identified within each image and the total number of cells in the region of interest (ROI) was counted. The average cell cross-sectional area was determined by dividing the total number of pixels within the ROI (consisting of nuclear and cytoplasmic pixels) by the number of cells in the ROI and converting the area from pixels to . Next, each image was thresholded to assign pixels within the nucleus to 0 and pixels within the cytoplasm to 1 for quantification of the number of nuclear pixels and cytoplasmic pixels. The NCR was calculated by dividing the number of nuclear pixels by cytoplasmic pixels. This analysis was done in ImageJ (NIH; http://rsbweb.nih.gov/ij/). 2.5.Quantification of the Optical Redox RatioFluorescence intensity images were constructed from the fluorescence lifetime images by integrating over time, summing all photons for each pixel. Any regions of noncellular structures, such as wrinkles within the tissue, were excluded from analysis. Additionally, a threshold was applied to isolate fluorescence from the cytoplasm and exclude nuclear fluorescence. The redox ratio was computed for each pixel by dividing the NADH fluorescence intensity by the FAD fluorescence intensity. The mean redox ratio for the image was computed by averaging the redox ratios from all cells, excluding the nuclear region. For each hamster, the cultured biopsy and frozen biopsy redox ratio measurements were normalized to the matched in vivo measurement to account for inter-animal and system variation. This analysis was done in Matlab (Natick, Massachusetts). 2.6.Quantification of the Fluorescence LifetimeA commercial software program, SPCImage (Becker and Hickl), was utilized to analyze the fluorescence decay data. In SPCImage, the measured system response of the second harmonic generation of urea crystals was de-convolved from the data. The resulting decay curve was then fit to the following model: where is the fluorescence intensity at time after the excitation pulse, and are the fractional contributions of the first and second emitting species (i.e., ), and are the fluorescence lifetimes of the first and second emitting species, and accounts for background light. Within the SPCImage software, after a threshold was set to isolate cytoplasmic fluorescence, the fluorescence decay for each pixel was evaluated. Then, matrices of , , , and were exported for further analysis. The mean , , , and for each image were averaged across all pixels in Matlab. Additionally, the mean fluorescence lifetime () was computed as a weighted average of and .2.7.Statistical AnalysisA Wilcoxon rank sum test was used to test for differences between the in vivo mean values of the redox ratio, NADH and FAD , , , and ratio and each cultured biopsy time point. Likewise, a Wilcoxon rank sum test was used to test for significance between optical measurements from in vivo and frozen-thawed tissue. For all tests, an level indicates significance. 2.8.Histological AnalysisHamster cheek pouch biopsies were collected and placed in chilled tissue media and refrigerated for 4, 8, 12, 24, or 48 h. The biopsy was placed in buffered formalin, paraffin embedded, sliced, and stained for Ki67 (a marker of proliferation) and cleaved caspase-3 (a marker of apoptotic cells). Traditional hematoxylin and eosin (H&E) staining was also performed to help visualize the epithelial layer. Ki67 and cleaved caspase-3 staining protocols were verified in positive control samples of the mouse small intestine and mouse thymus, respectively (data not shown). 3.ResultsOptical redox ratio values and NADH and FAD fluorescence lifetime values were acquired from the cheek pouch epithelium of 16 hamsters in vivo, from a cultured biopsy, and from a frozen-thawed biopsy. Figure 1 shows representative images of the normalized redox ratio, mean NADH lifetime, and mean FAD lifetime from the spinosum and basal layers of the epithelium in vivo, cultured tissue at 4, 12, 24, and 48 h after excision, and frozen-thawed tissue for the normal hamster epithelium. The redox ratio of the in vivo tissue appears to be similar to the cultured biopsy at 4 h and less than the cultured biopsy at 24 h. The frozen-thawed tissue appears to have a lower redox ratio than the in vivo tissue (Fig. 1). For the mean lifetimes of both NADH and FAD, the cultured tissue appears to have a lifetime similar to that of the in vivo tissue after 4 h; however, the frozen tissue shows an increased mean NADH lifetime relative to in vivo values (Fig. 1). No significant changes in cellular morphology are observed in the cultured or frozen tissue relative to the in vivo tissue measurements. Fig. 1Representative redox ratio (first row), NADH (second row), and FAD (third row) images from in vivo hamster epithelium at the basal (first column) and spinosum (second column) regions, tissue maintained in live culture 4, 12, 24, and 48 h after excision (third through sixth columns), and frozen-thawed tissue (last column). Each image is . is the mean lifetime ().  To compare deviations from the in vivo optical redox ratio in the cultured and frozen-thawed tissue samples, the optical redox ratio was normalized to the in vivo measurement. The variance in the redox ratio among in vivo measurements from the same hamster is 0.053, corresponding to a coefficient of variation (COV) of 8%. Figure 2 shows the normalized redox ratio of the in vivo tissue, cultured tissue over 4 h, and frozen-thawed tissue. No significant changes () in the redox ratio are observed at any time point for the 4 h the cultured biopsy was imaged [Fig. 2(a)]. The mean redox ratio of the cultured biopsy up to 4 h after excision is within 8% of the in vivo value. The redox ratio of the frozen-thawed sample, however, is 85% of the in vivo tissue measurement, and this difference is significant (). Fig. 2(a) The redox ratio value of frozen-thawed (F) hamster epithelium is significantly reduced from the in vivo (IV) value, while live tissue culture maintains redox ratio values similar to in vivo measures over the 4 h of live culture measurement ( of measurements). (b) The redox ratios of the spinosum and basal epithelial layers of the live-culture biopsies over 48 h ( of ). Asterisk denotes significant difference between in vivo and the indicated measurement (; ). 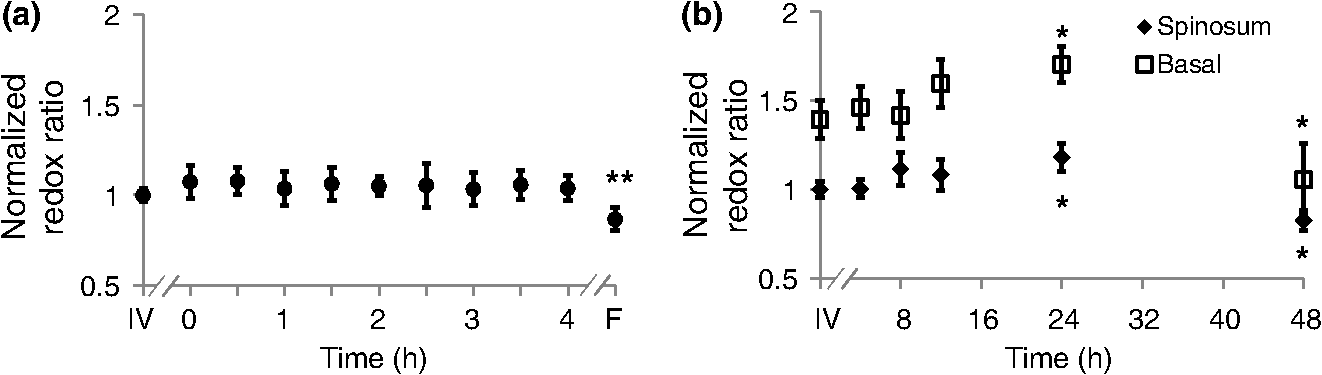 The metabolic activity of the stratified squamous epithelium is known to change with depth, with the most metabolically active cells occupying the basal layer of the epithelium.28 Therefore, an analysis of the metabolic change in multiple epithelial layers was evaluated over time. The redox ratio of the basal region is greater than that of the spinosum region at all time points [, Fig. 2(b)]. No significant change in the redox ratio is observed with time in live culture until 24 h, when the redox ratio of both the spinosum and basal cells increases [, Fig. 2(b)]. By 48 h, the redox ratio in both regions decreases [, Fig. 2(b)]. Quantitative parameters () of NADH and FAD fluorescence lifetimes (, , , and ) for the in vivo images are presented in Table 1. The mean lifetime () is a weighted average of the short () and long lifetime () components. Changes in hamster epithelium NADH fluorescence lifetime components relative to the in vivo values are shown in Fig. 3(a). Over the first 4 h, all of the mean NADH values from the live cultured biopsy are within 2.5% of the in vivo value (), while the frozen-thawed NADH is of the in vivo value, which is a statistically significant difference [; Fig. 3(a)]. Similarly, for NADH , all cultured biopsy measurements from the first 4 h are within 2% of the in vivo value (), while the frozen-thawed is [; Fig. 3(a)]. For NADH , the cultured biopsy values are within 1% of the in vivo NADH value () and the frozen-thawed mean is [; Fig. 3(a)]. The 4-h cultured biopsy NADH measurements are within 10% of the in vivo ratio (), while the frozen-thawed biopsy mean is 81% of the in vivo value [; Fig. 3(a)]. Fig. 3Changes ( of measurements) relative to in vivo (IV) values of NADH (a) and FAD (b) (diamond), (square), (triangle), and (cross) observed in tissues maintained in live culture for 4 h, and in frozen-thawed (F) tissues. is mean lifetime (). Asterisk denotes significant difference between in vivo and indicated measurement (; ; ).  Table 1Mean (SE) for in vivo measurements of NADH and FAD lifetime components (n=10).
τm is the mean lifetime (τ1*α1+τ2*α2). Similar trends are observed in the FAD lifetime values [Fig. 3(b)]. Over the first 4 h, the mean FAD of the cultured biopsy increases slightly but stays within 8% of the in vivo FAD [; Fig. 3(b)]. The frozen-thawed FAD is 9.5% greater than the in vivo FAD [; Fig. 3(b)]. Changes in FAD are within 6% for the cultured biopsy measurement and frozen-thawed samples [; Fig. 3(b)]. Likewise, the FAD values for the cultured and frozen-thawed samples vary less than 4% from the in vivo FAD [; Fig. 3(b)]. While no significant difference is observed in the FAD ratio between the 4-h cultured biopsy and the in vivo FAD (), the frozen-thawed measurement is 83% of the in vivo FAD ratio [; Fig. 3(b)]. When the cultured biopsy is assessed past 4 h, significant changes in the NADH fluorescence lifetime components are observed (Fig. 4). Both the spinosum and basal layers follow the same trends. By 12 h, NADH significantly decreases by 10% and remains significantly lower than the in vivo value out to 48 h in both the spinosum and basal layers [; Fig. 4(a)]. Again, at 12 h after excision, NADH and significantly decrease in both tissue layers [; Fig. 4(b) and 4(c)]. NADH is increased from the in vivo measurement at 12, 24, and 48 h after excision [; Fig. 4(d)]. Fig. 4Changes ( of measurements) relative to in vivo (IV) values of NADH (a), (b), (c), and (d) observed from both the spinosum (diamond) and basal (square) epithelial layers of tissues maintained in live culture for 48 h. is mean lifetime (). Asterisk denotes significant difference between in vivo and indicated measurement (; ).  Changes in the FAD lifetime of the spinosum layer are observed in the cultured biopsy at 48 h, when the FAD is significantly increased over the in vivo value [; Fig. 5(a)]. Both FAD () and () of the spinosum also increase at 48 h [Fig. 5(b) and 5(c)]. FAD decreased at 48 h in both the spinosum () and basal cells [; Fig. 5(d)]. Fig. 5Changes ( of measurements) relative to in vivo (IV) values of FAD (a), (b), (c), and (d) observed from both the spinosum (diamond) and basal (square) epithelial layers of tissues maintained in live culture for 48 h. is mean lifetime (). Asterisk denotes significant difference between in vivo and indicated measurement (, ).  Cellular morphology is maintained across the tissues regardless of preservation method (Fig. 6). No significant change () is measured in the average cellular cross-sectional area between the in vivo tissue, cultured biopsy, and frozen biopsy [Fig. 6(a) and 6(b)]. Likewise, the NCR remains unchanged () among the in vivo tissue, cultured biopsy, and frozen biopsy [Fig. 6(c) and 6(d)]. Fig. 6Cellular morphology, as measured by cell cross-sectional area [(a) and (b)] and nuclear-to-cytoplasmic ratio [(c) and (d)] remains unchanged () from in vivo (IV) values within both the spinosum and basal epithelial layers of tissues maintained in live culture over 48 h and in frozen-thawed (F) tissues ( of [ for (b) and (d)] measurements of a minimum of 20 cells per image). 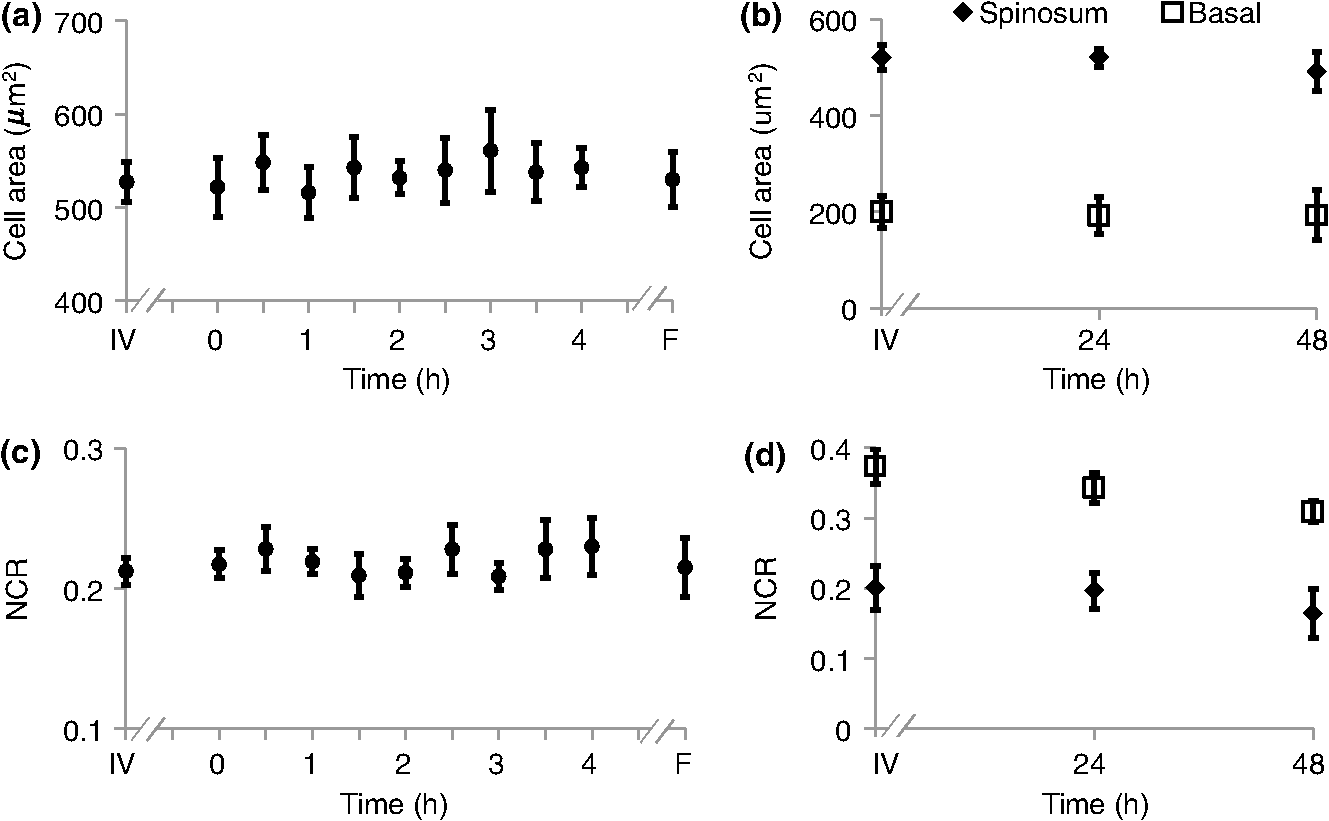 Histological analysis further confirms that the live tissue culture method maintains tissue viability. H&E stains reveal similar structure in the epithelial layer of the tissue at 4, 12, 24, and 48 h after excision (Fig. 7). Positive staining with antibodies against Ki67 reveals proliferating cells in the basal layer at all time points (Fig. 7). Furthermore, no apoptotic cells are observed in cleaved-caspase 3 stains of the epithelium at any time point (Fig. 7). Together, the histological analysis suggests that the cells of the hamster epithelium remain alive in tissue culture for 48 h after excision. Fig. 7Representative histology of the hamster epithelium. Hematoxylin and eosin staining demonstrates an intact epithelium (first row). Nuclear Ki67 staining (brown) of the basal cells suggests cellular proliferation at each time point (middle row). An absence of staining by anti-cleaved caspase-3 suggests no apoptosis (third row). Images were acquired using a air objective. 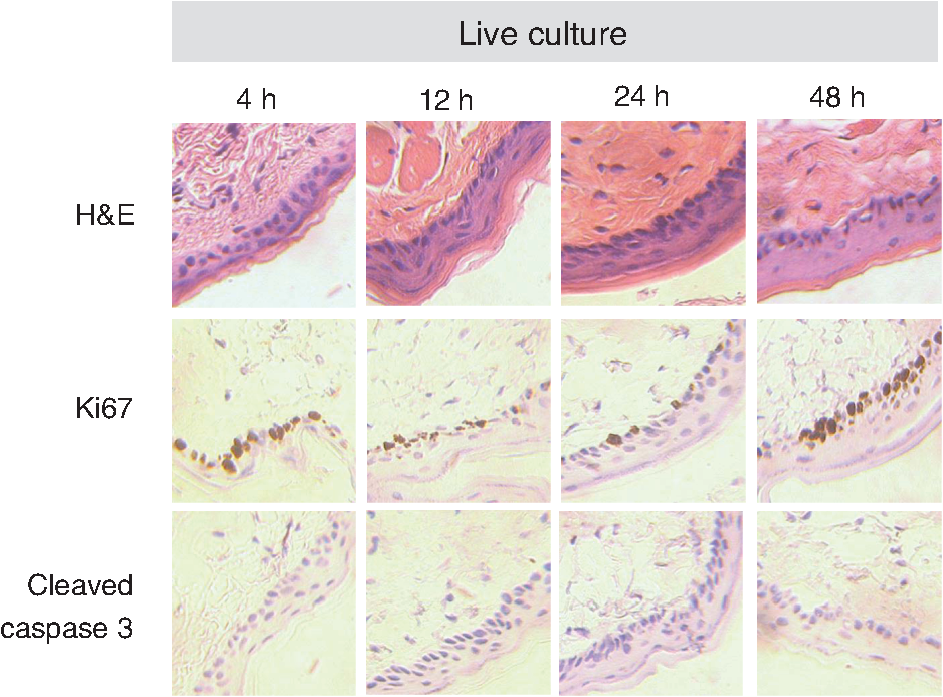 4.DiscussionThe goal of this study is to determine whether optical metabolic imaging of ex vivo tissues maintained in live culture reflects in vivo measurements. Optical endpoints include the optical redox ratio, NADH fluorescence lifetimes, and FAD fluorescence lifetimes, which are measured from in vivo tissue, a biopsy maintained in chilled tissue media, and a flash-frozen and thawed biopsy. Minimal changes, within 10% for the redox ratio and within 8% for the lifetime values, were observed between the in vivo hamster epithelium and the cultured tissue through 8 h after excision. However, statistically significant changes were observed in the frozen-thawed tissue. These results suggest that optical metabolism measurements from cultured tissue within 8 h after excision represent the in vivo metabolic state of the tissue, especially when compared to frozen-thawed tissue. Live tissue culture is used in many applications, including optical metabolic imaging and biochemical metabolic studies, to maintain viability of excised tissue.23,29–31 However, the temporal impact of ex vivo culture on the metabolic state of tissues has not been quantitatively assessed previously. Often, studies use the tissue for several days30 rather than the few hours investigated here. Cell viability studies have shown that for epithelial tissue, less than 2% cell death occurs over the first 5 days.30 However, no previous studies have confirmed that this live tissue culture approach provides optical metabolic measurements that are consistent with in vivo values, as we have shown in the current study. This live tissue culture approach is particularly attractive for optical imaging because these methods interrogate superficial layers of tissue ( for two-photon microscopy, depending on tissue type), so that oxygen and nutrients can diffuse into the tissue and maintain viability.32,33 These superficial interrogation volumes also avoid the necrotic and hypoxic cores that are found in some live tissue culture preparations.34 The optical redox ratio has been previously used to quantify metabolic differences among normal tissue, pre-cancerous tissue, and cancerous tumors.1,2,26,35,36 In the current study, two epithelial depth layers were interrogated because stratified squamous epithelial tissues are known to have a gradient of cellular metabolic activity, with the basal cells being most active and the superficial cells being least active.2,28 As shown in Fig. 2(b), the redox ratio of the basal region is greater () than the spinous region in vivo and at every time point during the live culture experiment, as expected. Additionally, Fig. 2(a) suggests that the optical redox ratio of frozen-thawed tissue does not completely describe the in vivo metabolic state. While the optical redox ratio is significantly reduced in frozen-thawed samples (), no significant difference is observed in the cultured tissue until 24 h after biopsy [Fig. 2(b)]. The change in redox ratio in the frozen-thawed tissue is due to both a decrease in the NADH fluorescence intensity and an increase in FAD fluorescence intensity (data not shown), which is consistent with decreased NADH fluorescence intensity observed between fresh and frozen-thawed hamster and cervical tissues.25,31 The NADH and FAD fluorescence intensities did not change significantly in the live tissue culture over 4 h (). The mean NADH lifetime of the hamster cheek epithelium is 1.149 ns (Table 1), which is consistent with previously reported NADH lifetime values.2,6,10,37,38 The mean NADH fluorescence lifetime is the averaged effect of the free and protein-bound lifetime components.2,11 While only minimal changes are observed in the NADH lifetime components of the cultured tissue until 12 h after biopsy, the frozen-thawed tissue has an increased , , and and a decreased ratio (Figs. 3 and 4). These results indicate that the fluorescence lifetimes of both the bound and free NADH are increased in the frozen-thawed tissue and that a greater portion of NADH exists in the bound conformation. Cells undergoing stress, such as apoptosis, have also shown increased mean NADH fluorescence lifetimes and increased proportions of bound NADH, suggesting that the freeze-thaw process exerts more stress than the live culture technique.37 Likewise, the observed FAD mean lifetime of 0.829 ns (Table 1) is similar to reported FAD lifetime measurements.2,39 For the FAD lifetime components, the frozen-thawed tissue has an increased and a decreased (Fig. 3). The decreased ratio indicates an increase in the relative amount of free (long-lifetime) FAD. This shift in relative amounts of the free and bound lifetime contributions explains the increased FAD . No significant change is observed in the FAD and , so the FAD fluorescence lifetime may be less sensitive to the cellular stress involved in the freeze-thaw process than the NADH fluorescence lifetime. Consistent cellular morphology across the in vivo, cultured, and frozen-thawed tissue indicates maintenance of tissue structure. The average cell cross-sectional areas observed in the hamster epithelial spinosum and basal layers are 530 and , respectively [Fig. 6(a) and 6(b)], which are consistent with published cell cross-sectional areas of hamster epithelium.40 Likewise, the observed NCR of the spinosum and basal layers of 0.2 and 0.4 [Fig. 6(c) and 6(d)], respectively, is consistent with previous reports.40,41 The current study is also consistent with previous studies reporting that cell size and NCR do not change in frozen-thawed cells/tissues or in live tissue culture relative to in vivo values.41,42 While the optical metabolic measurements varied from the in vivo values after 8 h in live culture, cellular morphology and histological analysis revealed no changes in cellular morphology, proliferation, or cell death over the 48-h time-course (Figs. 6 and 7). Therefore, optical metabolic imaging endpoints may be more sensitive to subcellular molecular changes than the histological or morphological analyses. Previous studies of the live tissue culture method have used histology to verify cell viability;30,33 however, our results suggest that molecular metabolic changes occur before changes in cellular proliferation or cell death. Thus, optical metabolic imaging may be a more sensitive endpoint than histology for detecting changes in cell status. To validate live tissue culture as an effective protocol for maintaining in vivo metabolic characteristics in excised tissue, the optical redox ratio, NADH fluorescence lifetime, and FAD fluorescence lifetime were quantified from hamster cheek epithelia in vivo, in live cultured biopsies followed for 48 h, and in frozen-thawed samples. The live tissue culture approach resulted in no significant change in any optical endpoint relative to in vivo measures until 12 h, when a decreased NADH was observed. Several significant differences, including a reduced redox ratio, increased NADH , and increased FAD were observed in the frozen-thawed samples. These results indicate that the live tissue culture method represents the in vivo state more accurately than the frozen-thawed procedure. Therefore, when in vivo optical measurements are not feasible, excised tissue may be maintained in chilled tissue media up to 12 h for close representation of in vivo metabolic states. AcknowledgmentsFunding sources include the NCI SPORE in Breast Cancer (P50 CA098131) and Vanderbilt University Provost Graduate Fellowship. ReferencesJ. H. Ostranderet al.,
“Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status,”
Cancer Res., 70
(11), 4759
–4766
(2010). http://dx.doi.org/10.1158/0008-5472.CAN-09-2572 CNREA8 0008-5472 Google Scholar
M. C. Skalaet al.,
“In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia,”
Proc. Natl. Acad. Sci. U.S.A., 104
(49), 19494
–19499
(2007). http://dx.doi.org/10.1073/pnas.0708425104 1091-6490 Google Scholar
W. L. RiceD. L. KaplanI. Georgakoudi,
“Two-photon microscopy for non-invasive, quantitative monitoring of stem cell differentiation,”
PLoS One, 5
(4), e10075
(2010). http://dx.doi.org/10.1371/journal.pone.0010075 1932-6203 Google Scholar
B. Chanceet al.,
“Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples: NADH and flavoprotein fluorescence signals,”
J. Biol. Chem., 254
(11), 4764
–4771
(1979). JBCHA3 0021-9258 Google Scholar
I. GeorgakoudiK. P. Quinn,
“Optical imaging using endogenous contrast to assess metabolic state,”
Annu. Rev. Biomed. Eng., 14 351
–367
(2012). http://dx.doi.org/10.1146/annurev-bioeng-071811-150108 ARBEF7 1523-9829 Google Scholar
D. K. Birdet al.,
“Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH,”
Cancer Res., 65
(19), 8766
–8773
(2005). http://dx.doi.org/10.1158/0008-5472.CAN-04-3922 CNREA8 0008-5472 Google Scholar
K. SuhlingP. M. FrenchD. Phillips,
“Time-resolved fluorescence microscopy,”
Photochem. Photobiol. Sci., 4
(1), 13
–22
(2005). http://dx.doi.org/10.1039/b412924p PPSHCB 1474-905X Google Scholar
N. P. Galletlyet al.,
“Fluorescence lifetime imaging distinguishes basal cell carcinoma from surrounding uninvolved skin,”
Br. J. Dermatol., 159
(1), 152
–161
(2008). http://dx.doi.org/10.1111/j.1365-2133.2008.08577.x BJDEAZ 1365-2133 Google Scholar
R. Patalayet al.,
“Quantification of cellular autofluorescence of human skin using multiphoton tomography and fluorescence lifetime imaging in two spectral detection channels,”
Biomed. Opt. Express, 2
(12), 3295
–3308
(2011). http://dx.doi.org/10.1364/BOE.2.003295 BOEICL 2156-7085 Google Scholar
M. C. Skalaet al.,
“In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia,”
J. Biomed. Opt., 12
(2), 024014
(2007). http://dx.doi.org/10.1117/1.2717503 JBOPFO 1083-3668 Google Scholar
J. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Publishers, New York
(1999). Google Scholar
J. R. Lakowiczet al.,
“Fluorescence lifetime imaging of free and protein-bound NADH,”
Proc. Natl. Acad. Sci. U.S.A., 89
(4), 1271
–1275
(1992). http://dx.doi.org/10.1073/pnas.89.4.1271 1091-6490 Google Scholar
F. TanakaN. TamaiI. Yamazaki,
“Picosecond-resolved fluorescence spectra of D-amino-acid oxidase: a new fluorescent species of the coenzyme,”
Biochemistry, 28
(10), 4259
–4262
(1989). http://dx.doi.org/10.1021/bi00436a021 MIRBD9 0144-0578 Google Scholar
P. P. ProvenzanoK. W. EliceiriP. J. Keely,
“Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment,”
Clin. Exp. Metastasis, 26
(4), 357
–370
(2009). http://dx.doi.org/10.1007/s10585-008-9204-0 CEXMD2 0262-0898 Google Scholar
M. W. Conklinet al.,
“Fluorescence lifetime imaging of endogenous fluorophores in histopathology sections reveals differences between normal and tumor epithelium in carcinoma in situ of the breast,”
Cell Biochem. Biophys., 53
(3), 145
–157
(2009). http://dx.doi.org/10.1007/s12013-009-9046-7 CBBIFV 1085-9195 Google Scholar
P. J. Tadrouset al.,
“Fluorescence lifetime imaging of unstained tissues: early results in human breast cancer,”
J. Pathol., 199
(3), 309
–317
(2003). http://dx.doi.org/10.1002/(ISSN)1096-9896 JPTLAS 0022-3417 Google Scholar
J. A. Jo,
“In vivo simultaneous morphological and biochemical optical imaging of oral epithelial cancer,”
IEEE Trans. Biomed. Eng., 57
(10), 2596
–2599
(2010). http://dx.doi.org/10.1109/TBME.2010.2060485 IEBEAX 0018-9294 Google Scholar
H. M. Chenet al.,
“Time-resolved autofluorescence spectroscopy for classifying normal and premalignant oral tissues,”
Laser Surg. Med., 37
(1), 37
–45
(2005). http://dx.doi.org/10.1002/(ISSN)1096-9101 LSMEDI 0196-8092 Google Scholar
P. A. De Beuleet al.,
“A hyperspectral fluorescence lifetime probe for skin cancer diagnosis,”
Rev. Sci. Instrum., 78
(12), 123101
(2007). http://dx.doi.org/10.1063/1.2818785 RSINAK 0034-6748 Google Scholar
K. M. Hansonet al.,
“Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient,”
Biophys. J., 83
(3), 1682
–1690
(2002). http://dx.doi.org/10.1016/S0006-3495(02)73936-2 BIOJAU 0006-3495 Google Scholar
J. Mizeretet al.,
“Endoscopic tissue characterization by frequency-domain fluorescence lifetime imaging (FD-FLIM),”
Lasers Med. Sci., 12
(3), 209
–217
(1997). http://dx.doi.org/10.1007/BF02765101 LMSCEZ 1435-604X Google Scholar
J. Requejo-Isidroet al.,
“High-speed wide-field time-gated endoscopic fluorescence-lifetime imaging,”
Opt. Lett., 29
(19), 2249
–2251
(2004). http://dx.doi.org/10.1364/OL.29.002249 OPLEDP 0146-9592 Google Scholar
R. Drezeket al.,
“Autofluorescence microscopy of fresh cervical-tissue sections reveals alterations in tissue biochemistry with dysplasia,”
Photochem. Photobiol., 73
(6), 636
–641
(2001). http://dx.doi.org/10.1562/0031-8655(2001)0730636AMOFCT2.0.CO2 PHCBAP 0031-8655 Google Scholar
I. Pavlovaet al.,
“Microanatomical and biochemical origins of normal and precancerous cervical autofluorescence using laser-scanning fluorescence confocal microscopy,”
Photochem. Photobiol., 77
(5), 550
–555
(2003). http://dx.doi.org/10.1562/0031-8655(2003)077<0550:MABOON>2.0.CO;2 PHCBAP 0031-8655 Google Scholar
G. M. Palmeret al.,
“Optimal methods for fluorescence and diffuse reflectance measurements of tissue biopsy samples,”
Lasers Surg. Med., 30
(3), 191
–200
(2002). http://dx.doi.org/10.1002/(ISSN)1096-9101 LSMEDI 0196-8092 Google Scholar
S. HuangA. A. HeikalW. W. Webb,
“Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein,”
Biophys. J., 82
(5), 2811
–2825
(2002). http://dx.doi.org/10.1016/S0006-3495(02)75621-X BIOJAU 0006-3495 Google Scholar
A. SchonleM. GlatzS. W. Hell,
“Four-dimensional multiphoton microscopy with time-correlated single-photon counting,”
Appl. Opt., 39
(34), 6306
–6311
(2000). http://dx.doi.org/10.1364/AO.39.006306 APOPAI 0003-6935 Google Scholar
B. Albertset al., Molecular Biology of the Cell, Garland Science, New York
(2002). Google Scholar
H. A. Krebs,
“Body size and tissue respiration,”
Biochim. Biophys. Acta., 4
(1–3), 249
–269
(1950). http://dx.doi.org/10.1016/0006-3002(50)90032-1 BBADEX 0925-4439 Google Scholar
O. A. Trowell,
“The culture of mature organs in a synthetic medium,”
Exp. Cell Res., 16
(1), 118
–147
(1959). http://dx.doi.org/10.1016/0014-4827(59)90201-0 ECREAL 0014-4827 Google Scholar
C. K. Brookneret al.,
“Autofluorescence patterns in short-term cultures of normal cervical tissue,”
Photochem. Photobiol., 71
(6), 730
–736
(2000). http://dx.doi.org/10.1562/0031-8655(2000)071<0730:APISTC>2.0.CO;2 PHCBAP 0031-8655 Google Scholar
P. T. Soet al.,
“Two-photon excitation fluorescence microscopy,”
Annu. Rev. Biomed. Eng., 2 399
–429
(2000). http://dx.doi.org/10.1146/annurev.bioeng.2.1.399 ARBEF7 1523-9829 Google Scholar
R. W. Verweret al.,
“Cells in human postmortem brain tissue slices remain alive for several weeks in culture,”
FASEB J., 16
(1), 54
–60
(2002). http://dx.doi.org/10.1096/fj.01-0504com FAJOEC 0892-6638 Google Scholar
S. M. Evanset al.,
“Detection of hypoxia in human squamous cell carcinoma by EF5 binding,”
Cancer Res., 60
(7), 2018
–2024
(2000). CNREA8 0008-5472 Google Scholar
H. N. Xuet al.,
“Quantitative mitochondrial redox imaging of breast cancer metastatic potential,”
J. Biomed. Opt., 15
(3), 036010
(2010). http://dx.doi.org/10.1117/1.3431714 JBOPFO 1083-3668 Google Scholar
Z. Zhanget al.,
“Metabolic imaging of tumors using intrinsic and extrinsic fluorescent markers,”
Biosens. Bioelectron., 20
(3), 643
–650
(2004). http://dx.doi.org/10.1016/j.bios.2004.03.034 BBIOE4 0956-5663 Google Scholar
H. W. Wanget al.,
“Differentiation of apoptosis from necrosis by dynamic changes of reduced nicotinamide adenine dinucleotide fluorescence lifetime in live cells,”
J. Biomed. Opt., 13
(5), 054011
(2008). http://dx.doi.org/10.1117/1.2975831 JBOPFO 1083-3668 Google Scholar
M. WakitaG. NishimuraM. Tamura,
“Some characteristics of the fluorescence lifetime of reduced pyridine nucleotides in isolated mitochondria, isolated hepatocytes, and perfused rat liver in situ,”
J. Biochem., 118
(6), 1151
–1160
(1995). JOBIAO 0021-924X Google Scholar
N. Nakashimaet al.,
“Picosecond fluorescence lifetime of the coenzyme of D-amino acid oxidase,”
J. Biol. Chem., 255
(11), 5261
–5263
(1980). JBCHA3 0021-9258 Google Scholar
J. W. EvesonD. G. MacDonald,
“Quantitative histological changes during early experimental carcinogenesis in the hamster cheek pouch,”
Br. J. Dermatol., 98
(6), 639
–644
(1978). http://dx.doi.org/10.1111/bjd.1978.98.issue-6 BJDEAZ 1365-2133 Google Scholar
F. H. WhiteK. Gohari,
“Cellular and nuclear volumetric alterations during differentiation of normal hamster cheek pouch epithelium,”
Arch. Dermatol. Res., 273
(3–4), 307
–318
(1982). http://dx.doi.org/10.1007/BF00409260 ADREDL 0340-3696 Google Scholar
T. S. HauschkaJ. T. MitchellD. J. Niederpruem,
“A reliable frozen tissue bank: viability and stability of 82 neoplastic and normal cell types after prolonged storage at ,”
Cancer Res., 19
(6, Pt 1), 643
–653
(1959). CNREA8 0008-5472 Google Scholar
|



