
|
|
1.IntroductionTwo-photon fluorescence microscopy1 is widely used to image biological tissues, especially at depth. This is due to the ability of near-infrared light to penetrate deeper tissue layers and induce two-photon fluorescence that allows high-resolution imaging at the cellular level.2–10 Biological tissue is an intrinsically turbid medium, with optical properties characterized by strong multiple scattering and heterogeneity in its refractive index. Excitation light that induces two-photon fluorescence at the focal area is attenuated by scattering and absorption and consequently imaging depth is limited. Light in biological tissue is strongly forward-scattered, and the optical properties of the media can be characterized by the absorption coefficient , the scattering coefficient , the anisotropy factor and the reduced scattering coefficient ().3,9 For most biological tissues the value of g is in the range of 0.6 to 0.95 and is in the range of 5 to .6,11,12 Some tissue components, such as blood and melanin,12,13 may have a noticeable absorption at visible or near infrared (IR) excitation wavelengths. However, the values of the absorption coefficients usually are an order of magnitude lower in comparison to those of the scattering coefficient, so that effectively, absorption has no influence on the achievable imaging depth.6,11 On the other hand, due to the high values of in turbid media, most of the light will be multiply scattered before reaching the focal area. The intensity of the unscattered (ballistic) photons effectively reaching the focal area decays exponentially with depth, while the total amount of light decays approximately as the inverse of depth.3 However, only unscattered photons can contribute to two-photon fluorescence, since the scattered photons will be time-delayed or miss the focal area. For this very reason, two-photon induced fluorescence is localized only in a small (micron scale) focal area inside the sample that allows high-resolution imaging in turbid media. Variations in the index of refraction along the excitation path, commonly encountered in tissue, also reduce the efficiency of two-photon excitation. Clearly, an increase in the excitation light power will deliver more photons to the focal area, and consequently increase the achievable imaging depths. However, as shown in Ref. 3, beyond a certain power level the excitation light will also induce out-of-focus fluorescence right below the sample surface, which will mask the fluorescence signal from the focal area and thus limit the achievable imaging depth. Most of the research performed to date has focused on the delivery of more excitation power to the focal area in an attempt to generate a more intense fluorescence signal at deeper layers. This strategy is counterproductive in imaging of biological samples because of the potential for photodamage to cells and surrounding tissues at the focal point. Imaging depth depends on the ability of the system to deliver sufficient excitation light power at the focal point to induce two-photon fluorescence, whose efficiency is proportional to the square of the peak power of the excitation light. Since the excitation light peak power is a function of pulse duration and energy, shorter pulses of higher energy are preferable for two-photon fluorescence imaging.14 Theer et al. showed that an imaging depth of 1 mm could be attained in brain tissue when a regenerative amplifier was employed to boost the excitation power.2 The numerical aperture (NA) of the focusing optics affects resolution as well as imaging depth; an NA of 0.6 to 0.8 has been reported as optimal to image at a depth of 300 μm in tissue phantoms.15 Since light with longer wavelengths is scattered less in turbid media, its use should increase the imaging depth. For example, Kobat et al.16 succeeded in imaging a mouse brain to 1 mm depth with an excitation wavelength of 1280 nm. The application of optical clearing agents,17 which actually are index-matching compounds, doubled the image depth in a skin tissue sample (from 40 to 80 μm). Another factor that hinders imaging depth in tissue is the optical inhomogeneity that leads to a distortion of the excitation light wavefront and degrades imaging performance. This problem can be partially corrected with the use of adaptive optics.18–20 Another major difficulty associated with fluorescence imaging at depth is the harvesting of fluorescence photons. During the acquisition of images in deep layers, the detected signal decays with depth due to the decrease in numbers of induced fluorescence photons, as well as fluorescence attenuation by multiple scattering and absorption processes. Evidently, the ability of the imaging system to collect fluorescence photons that propagate in the turbid sample will strongly influence the imaging depth, and thus more sensitive fluorescence detectors are needed to image deeper layers. The overall sensitivity also relies on the efficiency of the optical configuration for fluorescence collection. All commercial and experimental two-photon fluorescence microscope systems utilize an optical design, where fluorescence is collected by the same microscope objective used for excitation. The collection efficiency of this method is greatly limited by the ability of the microscope objective to collect photons only at a certain angle and from a relatively small area of the sample, thus leaving most of the fluorescence photons undetected. One strategy, termed total emission detection (TED), uses a parabolic mirror to reflect the “escaped” photons back to the detector and increase the signal/noise gain (roughly ) and imaging performance.21,22 Alternatively, optical fibers can be used for fluorescence collection in imaging systems.23 The relatively narrow acceptance angle of optical fibers limits their ability to collect fluorescence photons. However, the use of an array of optical fibers, especially large-core fibers with high , can prove efficient. The additional harvesting of fluorescence photons by a ring of large-core optical fibers surrounding the objective was shown to potentially enhance the overall fluorescence signal collection efficiency by up to four-fold.24 To solve the problem associated with the loss of fluorescence photons in conventional fluorescence microscopy (described above), we separated the excitation and detection optics to allow a more efficient collection of fluorescence and enhancement of the achievable imaging depth.25,26 The excitation optics retain their conventional design, however, our detection method introduces several innovative approaches. Our experimental results on deep imaging of turbid samples with optical properties that simulate brain tissue in addition to in vivo and ex vivo imaging of various biological tissues attest to the advantages of this approach. To the best of our knowledge, the reported imaging depth in brain tissue does not exceed a few hundred microns,6,8,27 with a maximum reported range of about 1 mm.2,16 The use of the detection method described below allowed us to perform imaging in turbid samples with brain-like optical properties (, )11,28 at depths up to 3 mm, while the maximum depth at which images could be acquired on the same samples by the state-of-the-art commercial two-photon microscope (Zeiss LSM 710) was limited to about 500 μm. Although the presented experimental setup utilizes an optical scheme where excitation and detection are performed on the opposite sides of the sample, we have previously presented a possible configuration in which excitation and detection optics are combined in the same unit to perform imaging from the same side of the sample (reflectance geometry).25 We have modified our deep-tissue imaging system, described below, to adapt it for the in vivo imaging of small animals. Using this system we were able to successfully obtain high resolution fluorescence images of both intrinsic contrast and of stem cells in the small intestine and colon of transgenic mice expressing eGFP from the Lgr5 promoter in intestinal stem cells.29 Although recent work has characterized intrinsic contrast in healthy and diseased gastric tissue, the current conventional two-photon systems are limited to a depth of 170 μm in gastric tissue.30 Here we image and discriminate tissue structures, intrinsic contrast and single cells to a depth of up to 420 μm in living tissue. The murine small intestine has become an important in vivo model for studying stem cell renewal and cell fate decisions, mucosal immunology, and intestinal carcinogenesis, due the high proliferative rate of epithelial cells. The status and function of stem cells is strictly regulated; one major regulator of cell proliferation, differentiation and stem cell self-renewal in the intestinal crypt is the Wnt signaling pathway.30–33 The Wnt target gene encoding Lgr5 (leucine-rich-repeat-containing-G-protein-coupled receptor 5, also known as Gpr49) has been recently identified as a multi-potent stem cell marker located in the crypt bases of the intestinal epithelium.29 Tightly regulated self-renewal mediated by Wnt signaling in stem cells and progenitor cells is subverted in cancer cells leading to malignant proliferation. Deletion of the adenomatous polyposis coli (APC) gene, a tumor suppressor, in the stem cells has been shown to drive aberrantly high levels of Wnt signaling leading to malignant transformation within the small intestine and colon.34 The ability to resolve and track single stem cells would provide important insights into the regulation of the position and number of stem cells to maintain tissue homeostasis. Our deep imaging set-up allows us to image through the entire thickness of the small intestine and colon epithelia, from the undifferentiated crypt base, through the rapidly proliferating “committed progenitor” compartment, to terminally differentiated cells on the villus surface. The possibility of imaging the same epithelial cell through its differentiation path would provide important information on the cell’s behavior and physiology in its native microenvironment. 2.Materials and Methods2.1.Turbid Sample PreparationTurbid samples made of silicone resin and particles (Atlantic Equipment Engineers, Ti-602) as a scattering agent, were prepared according to Ref. 28 to which fluorescent microspheres (Invitrogen FluoSpheres yellow-green, size 1 to 15 μm) were added. These samples were polymerized in petri dishes to obtain a sample size of . The reduced scattering and absorption coefficients of the samples were measured at 806 nm as 10 and , respectively, by the method described in Ref. 28. Turbid samples were also prepared by dispersion of fluorescent microspheres and particles in gelatin and agar matrices in place of the silicone resin.25 The two types of samples have the same optical properties, but while the agar/gelatin samples are easier to prepare, the silicone resin samples do not degrade and thus can be used for longer periods of time. 2.2.Specimen PreparationFor in vivo imaging, four to six-week-old mice were fasted for 24 h prior to being anesthetized with 0.3 cc of ketamine-xylazine given intraperitoneal (IP), and immobilized (with clear tape) on their side on a glass slide. A 1-cm vertical incision was made in the lower left quadrant of the abdomen and a loop of distal small bowel exteriorized. The loop was placed on the surface of the glass slide, with a minimal volume of phosphate buffered saline (PBS) to prevent desiccation of the serosal surface. The prepared animal was placed on the stage of the multiphoton microscope and imaged. At the end of the imaging session, the animal was euthanized. For ex vivo tissue imaging, the mouse is euthanized. Freshly excised small intestinal tissue is rinsed with PBS and imaged. All imaging occurred within 4 h of euthanasia. All procedures were reviewed and approved by the UC Irvine Institutional Animal Care and Use Committee (protocol 2002-2357). 2.3.Two-Photon Microscope SystemThe principles of operation and characteristics of our two-photon microscope system and its ability to image inside turbid media up to a depth of a few mm have been previously described.25,26 Here we present a modified upright version of the previous model that is better adapted to image live small animals. The experimental system diagram is shown in Fig. 1. A femtosecond Ti:Sapphire laser (Mai Tai, Spectra-Physics) supplied with a group velocity dispersion (GVD) compensation attachment (DeepSee) is used for the two-photon excitation of the sample. The excitation beam is directed to an acousto-optic modulator (AOM, AA Opto-Electronics MT 110-B50A1) to regulate the power level. Afterward, the beam is passed through an --scanner (ISS, Model 6220) coupled to an Olympus BX illumination module equipped with a long working distance (7-mm) Olympus LCPlanFl air objective. Focusing and positioning of the sample is achieved via a motorized -- stage. The two-photon fluorescence is induced by the IR beam focused inside the sample from its top side and collected by the detector (described below) from the opposite side of the sample. In order to protect the extremely sensitive detector from external light, the components are enclosed in a black, light-tight box. Fig. 1Schematic diagram of the deep-tissue imaging system. A dispersion compensated Maitai DeepSee laser is attenuated by an acoustic-optic modulator (AOM) and delivered to the sample using a galvano-scanner. The detector is placed in the transmission path of the Olympus BX71 upright microscope. 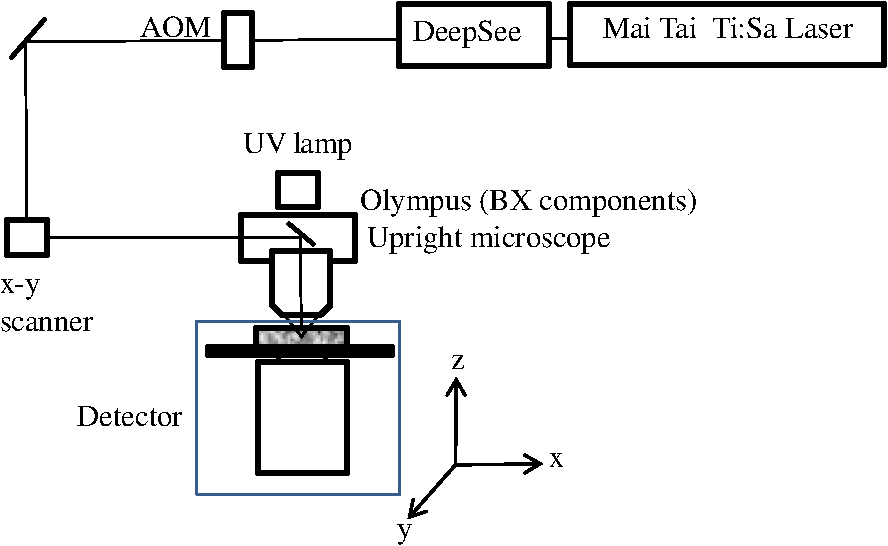 The detector is the key component of the experimental setup described above. The detector and the principle of its operation are shown schematically in Fig. 2(a). The detector is composed of a head-on photomultiplier tube (PMT) with a wide area photocathode (Hamamatsu PMT R1104, diameter 25 mm, or R7600P-300, ) operating in photon-counting mode. The R1104 PMT is attached to a glass cylinder with mirror-coated walls, which in turn acts as a light-guide by directing fluorescence photons to the PMT photocathode. The R7600P-300 PMT is coupled directly to the optical shutter window without use of light-guide cylinder. The optical shutter is made of two optical filters (Schott BG-39) that transmit fluorescence, but prevent the excitation light from entering the PMT. A gap between the filters allows the introduction of a thin 0.1-mm aluminum plate to provide a mechanical shutter. All the detector components, including the gap between the filters, are immersed in a refractive index matching fluid to curtail losses of fluorescence photons due to reflection at the boundaries. For the same reason, during measurements, the detector assembly is placed directly in contact with the surface of the sample. Fig. 2(a) Fluorescence detection method: developed in this work showing the transmission geometry. (b) The conventional reflectance geometry found in most commercial two-photon fluorescence microscopes. 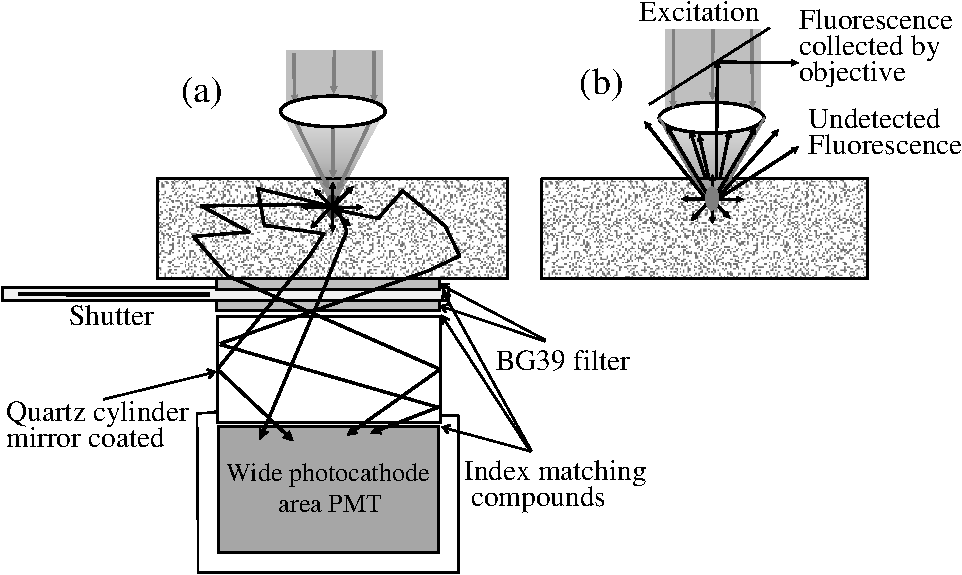 As shown in Fig. 2(a), the two-photon-induced fluorescence is multiply scattered into the turbid sample and collected by the detector. The multiple scattering aids detection, since photons initially headed away from the detector now have a chance to be redirected and enter the detector. For the same reason, in the multiple-scattering media, where photons constantly change direction, light losses due to total internal reflection at the sample/detector boundary are reduced and fluorescence photons can be captured by the detector from any angle of incidence. In theory, multiple scattering could additionally improve fluorescence detection, since in multiple-scattering media the intensity of the scattered photons decays as the inverse of the distance from the excitation light focus,3 which is less than the inverse of the squared distance decay in clear media. Our detection method has proven to be very efficient in harvesting photons, as indeed no microscope objective has the ability to collect photons from such a wide area.25,26 However, we acknowledge that our detection geometry does not capture the epi-fluorescence photons, which comprise a fraction of the total fluorescence photons under conditions of deep-tissue two-photon excitation. The diagram in Fig. 2(b) illustrates the principle of operation of a conventional two-photon microscope, where fluorescence photons can only be collected by a microscope objective from a relatively small area and at a narrow angle. We have also used a commercial two-photon scanning microscope Zeiss LSM 710 to compare results obtained with our instrument. 3.Results and Discussion3.1.Phantom StudiesFigure 3 presents images of fluorescent beads dispersed in turbid samples (1-cm thickness) and acquired at various depths. Similar results were obtained with samples made of gelatin or agar gel and silicone. Using our fluorescence detection method we were able to image fluorescent beads in turbid samples at depths up to 3 mm [Fig. 3(a)].The same imaging experiment was conducted with a commercial Zeiss LSM 710 two-photon fluorescence microscope with similar objective and excitation light power, obtaining a maximum imaging depth of about 0.5 mm [see Fig. 3(a)]. It is important to note that 0.5 mm is indeed the maximum imaging depth achieved so far in brain tissue,6,8,27 and the turbid samples we used for imaging had brain-like optical properties (, ). Fig. 3Images of fluorescent beads in turbid samples: comparison of imaging depths. (a) 1 μm beads imaged at 200 μm and 450 μm depth obtained with a Zeiss LSM710 with multiphoton optics and using our system at depths of 670, 1090, 1830, 2020, 2630, and 2980 μm, respectively. (b) Detail of an Image of 15 μm beads at 1.83 mm depth. (c) images of two 2 μm beads at 2.23 mm depth (c).  To confirm that our system maintains a high resolution of images at increased depths we prepared turbid samples containing a mixture of 2-μm and 15-μm fluorescent beads. Figure 3(b) shows the image of a 15-μm bead at a depth of 1.83 mm. Figure 3(c) shows two close 2-μm fluorescent beads also at a depth of 2.23 mm. The two beads appear well resolved at this depth and their apparent sizes correspond to the expected ones. The images depicted in Fig. 3 were acquired using an objective with (Olympus LCPlanFl ) for excitation. Because the beam diameter at the focus depends on the numerical aperture, the use of higher NA objectives results in a smaller beam size, which in turn gives a higher excitation light intensity at the focus with an increased imaging depth and resolution. The drawback is that high NA objectives usually have a short working distance. It was shown in Ref. 15 that objectives with an NA of 0.6 to 0.8 are the best choice for two-photon imaging in turbid media. 3.2.Tissue ImagingOur system has been successfully employed in the imaging, both in vivo and ex vivo, of adult stem cells at the crypt basis of the murine small intestine and colon. Figure 4 shows the schematic diagram of colon and small intestine along with the imaging setup. In Figs. 5 and 6 the morphology of the normal colon and small intestine tissue is imaged with our two-photon fluorescence microscope. Fig. 4Schematic diagrams of (a) small intestine and (b) colon mucosa. (c) Imaging scheme: images were acquired from the outside of the intact gastric lumen; villus (I), crypt (II), lamina propria (III), collagen layer (IV), muscularis mucosa (V), submucosa (VI), muscularis externa (VII, VIII), stem cell (IX), and Paneth cell (X).  Fig. 5Colon images of intrinsic contrast and crypts. Progressively deeper images reveal submucosal collagen, then cross sections of GFP-bright crypt bases (in some crypts) that transition into the transit-amplifying compartment and become more attenuated at depth. Field of view . Excitation wavelength 880 nm.  Fig. 6Small intestine images. White arrows correspond to collagen fibers at 30 μm depth and to individual at 70 μm depth. Field of view . Excitation wavelength 880 nm. 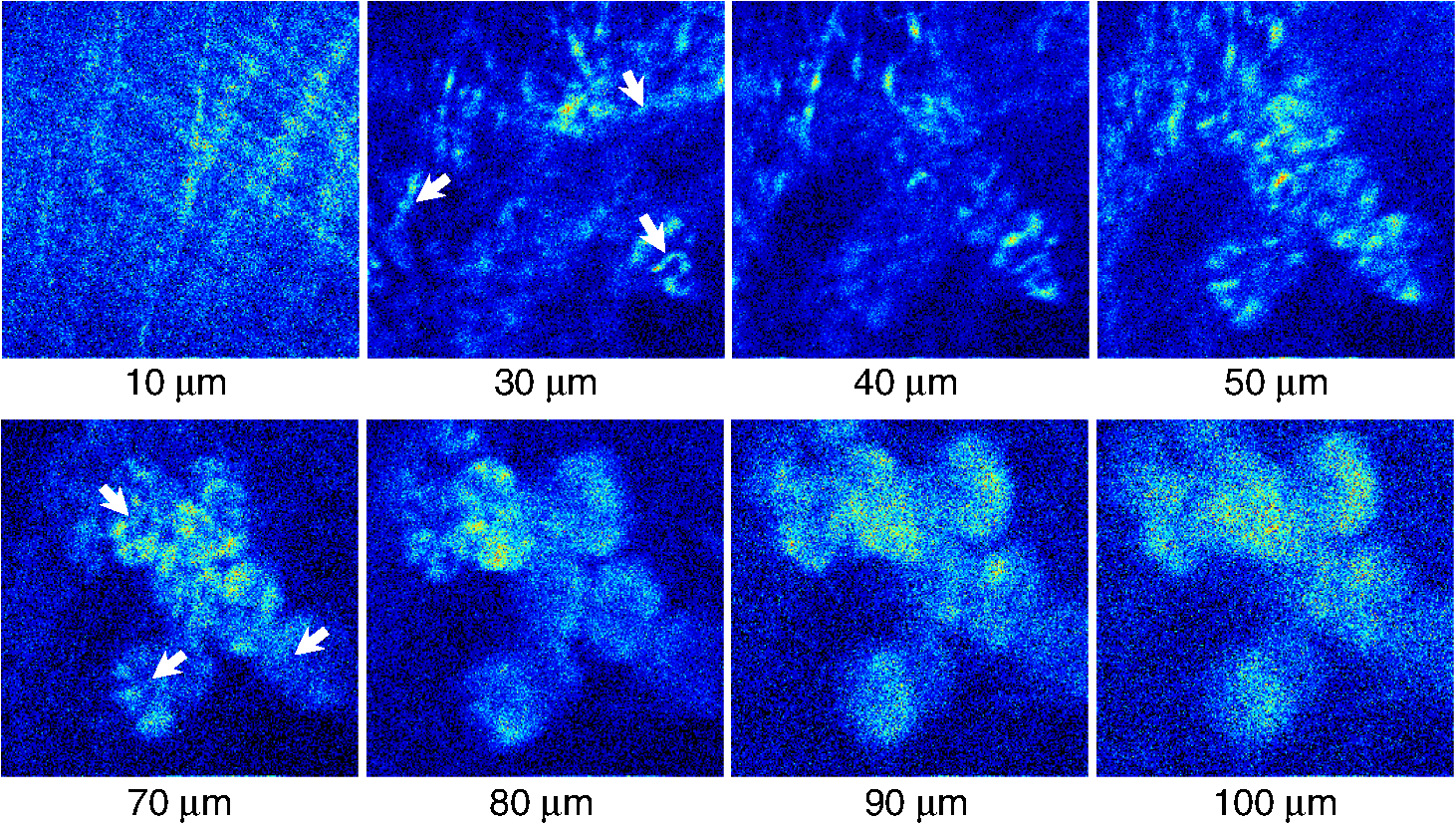 The deep two-photon imaging is performed with an excitation wavelength of 880 nm through the external muscularis mucosa [Fig. 4(c)] up to a depth of 420 μm (Fig. 5). The same sample imaged with the commercial Zeiss LSM 710 microscope could only be imaged up to a depth of 160 μm. Different structures can be discerned at different depths of the colon (Fig. 5) and small intestine (Fig. 6) tissue. Beginning from the surface of the colon tube we first imaged the muscularis externa, the muscularis mucosa, and the submucosa [Fig. 4(b)].35 In these layers, at 122 and 133 μm depths, the collagen walls of a branching blood vessel can be observed. At 175 and 195 μm, the beginning of the epithelium structure is marked by the presence of the bases of the circular crypts that contain the stem cells and are surrounded by collagen fibers. Single stem cells can be seen at 217 μm depth (Fig. 5) within the circular crypt because of their distinctive alternated pattern.29 Below a 217 μm depth, the granular level of the colon is visible with circular crypts and the lamina propria that contains the capillary network.35 The small intestine three-dimensional (3-D) structure presents similarities to the colon, but some differences can be observed.35 The surface of the small intestine exteriorized loop was the starting point from which we first imaged the smooth muscle layer, characterized by smooth muscle cells at depths of 10 and 20 μm. A layer of collagen fibers is located at the base of the Lgr5-GFP stem cells positive crypts at 30 μm depth. Again, single stem cells are visible at depth 70 μm within the circular crypt with a characteristic pattern of alternating fluorescent cells and nonfluorescent Paneth cells (Fig. 6).29 Imaging at different excitation wavelengths allows one to selectively investigate different intrinsic fluorophores, providing identification of different tissue components. Figure 7 shows the intrinsic contrast of the colon tissue at two different excitation wavelengths at the depth of 80 μm. In Fig. 7(a) at the 880 nm excitation wavelength two crypts are visible (white arrows) and blood capillaries and collagen are also visible from within the lamina propria. Two-photon fluorescence intensity of the same field of view excited at 740 nm [Fig. 7(b)] highlights the contrast of the metabolic coenzyme nicotinamide adenine dinucleotide (NADH) within single epithelial cells in the round crypt base. Subcellular details such as the dim nuclei and bright mitochondria are distinguishable. Since NADH is the principal electron acceptor in glycolysis and electron donor in oxidative phosphorylation it provides important information about single-cell metabolism.36 Fig. 7Two-photon fluorescence images of colon from a mouse excited at 880 nm (a) and 740 nm (b). Field of view , depth 80 μm.  Deep in vivo imaging of NADH and other fluorophores [Fig. 7(b)] in epithelial cells and myofibroblasts, vascular endothelia, and immune cells within the lamina propria [Fig. 7(a)] could provide important information about single-cell metabolism in the native microenvironment and about physiological processes related to inflammation and cancer in colon and small intestine. 3.3.Deep-Tissue Fluorescence ImagingA useful conceptual framework for considerations of fluorescence imaging deep into tissue or other highly scattering media is a separation of the excitation and fluorescence emission processes. Unquestionably, two-photon excitation achieves the highest resolution (microns) images and the deepest penetration depths. Increasing excitation laser power can achieve deeper penetration, but the cost in photodamage to tissue, especially live tissue, is unacceptable. In addition, high laser excitation fluxes induce significant spurious fluorescence signals at the sample surface. In our experience, we can obtain two-photon excitation at a depth of up to 3 mm in a scattering tissue (). The excitation efficiency degrades in more highly scattered or inhomogeneous (varying indices of refraction) tissue regions, as does the optical resolution. Significantly, in our transmission geometry the emitted fluorescence photons from the two-photon excitation focal volume can still be detected after traversing an additional of scattering media before arriving at the detector. Our success in high resolution (1 μm) imaging at depth in tissue (ex vivo or in vivo) is achieved through the combination of several factors that vastly increase the efficiency of the detection of fluorescence photons. Namely, these factors include a simplified optical path, a large area detector and importantly an index matching fluid for the optical path that minimizes photon loss. The other systems in the literature do not use these successful strategies.21–23 Our current microscope design relies on transmission geometry, rather than the conventional epi-fluorescence detection through the objective. While the conventional epi-configuration is versatile for whole animal or large-tissue imaging experiments, its deep imaging capabilities are generally constrained to the 100 to 500-μm range. It is noteworthy, that Combs et.al.23 have adapted their design for an epi-configuration (epiTED) for in vivo imaging of various tissue types (e.g., brain and kidney) with emission collection gains of two or more. Our group is currently adapting our design criteria to the epi-configuration.25 In our transmission geometry we used highly scattered samples of about 1 cm thick. However, the thickness limit is due to the absorption in the material rather than to the scattering properties. 4.ConclusionsIn this manuscript, we present a fluorescence detection method that allows one to obtain fluorescent images with micron scale resolution inside tissue-like turbid media. The detector collects photons from a wide area of the sample with minimum photon losses, which makes it exceptionally sensitive to low light levels. We showed that by using this detector it is possible to increase imaging depth of samples emulating tissue optical properties up to 3 mm, while the best state-of-the-art commercial two-photon fluorescence microscope was able to image the same samples only at a maximum depth of 0.5 mm. We performed in vivo deep two-photon microscopy on an exteriorized loop of intact, live mouse small intestine and freshly excised colon tube, using a transgenic line expressing eGFP in Lgr5 positive stem cells. We were able to image up to 420 μm deep and to discriminate and identify different important structures of the colon and small intestine tissue. Performing in vivo deep tissue imaging on an exteriorized loop of small intestine allows us to monitor the spatial and temporal dynamics of stem cells, intrinsic contrast and collagen organization. This type of in vivo microscopy can be used with any transgenic reporter mouse or fluorescently tagged cell line, making it useful for studying any number of normal or pathogenic developmental processes, alterations in cell metabolism, or carcinogenesis, with single-cell resolution. AcknowledgmentsThis work was supported by National Institutes of Health Grants: P41-RRO3155, 8P41GM103540, P50-GM076516 and ICTS: UL1 TR000153 and the Keck’s Foundation grant 44769549507. ReferencesW. DenkJ. StricklerW. Webb,
“Two-photon laser scanning fluorescence microscopy,”
Science, 248
(4951), 73
–76
(1990). http://dx.doi.org/10.1126/science.2321027 SCIEAS 0036-8075 Google Scholar
P. TheerM. T. HasanW. Denk,
“Two-photon imaging to a depth of 1000 micron in living brains by use of a Ti:Al2O3 regenerative amplifier,”
Opt. Lett., 28
(12), 1022
–1024
(2003). http://dx.doi.org/10.1364/OL.28.001022 OPLEDP 0146-9592 Google Scholar
P. TheerW. Denk,
“On the fundamental imaging-depth limit in two-photon microscopy,”
J. Opt. Soc. Am. A, 23
(12), 3139
–3149
(2006). http://dx.doi.org/10.1364/JOSAA.23.003139 JOAOD6 0740-3232 Google Scholar
D. O’Malley,
“Imaging in depth: controversies and opportunities,”
Biophysical Tools for Biologists: In Vivo Techniques (Methods in Cell Biology), 95
–128 Academic Press, London, United Kingdom
(2008). Google Scholar
M. GuX. GanA. KistemanM. G. Xu,
“Comparison of penetration depth between two-photon excitation and single-photon excitation in imaging through turbid tissue media,”
Appl. Phys. Lett., 77
(10), 1551
–1553
(2000). http://dx.doi.org/10.1063/1.1308059 APPLAB 0003-6951 Google Scholar
X. DengM. Gu,
“Penetration depth of single-, two-, and three-photon fluorescence microscopic imaging through human cortex structures: Monte Carlo simulation,”
Appl. Opt., 42
(16), 3321
–3329
(2003). http://dx.doi.org/10.1364/AO.42.003321 APOPAI 0003-6935 Google Scholar
H. C. GerritsenC. J. De Grauw,
“Imaging of optically thick specimen using two-photon excitation microscopy,”
Microsc. Res. Tech., 47
(3), 206
–209
(1999). http://dx.doi.org/10.1002/(SICI)1097-0029(19991101)47:3<>1.0.CO;2-1 MRTEEO 1059-910X Google Scholar
M. Oheimet al.,
“Two-photon microscopy in brain tissue: parameters influencing the imaging depth,”
Methods, 11
(1), 29
–37
(2001). http://dx.doi.org/10.1016/S0165-0270(01)00438-1 MTHDE9 1046-2023 Google Scholar
A. LerayC. OdinY. Le,
“Out-of-focus fluorescence collection in two-photon microscopy of scattering media,”
Opt. Commun., 281
(24), 6139
–6144
(2008). http://dx.doi.org/10.1016/j.optcom.2008.09.032 OPCOB8 0030-4018 Google Scholar
E. J. BotcherbyR. JuskaitisT. Wilson,
“Scanning two photon fluorescence microscopy with extended depth of field,”
Opt. Commun., 268
(2), 253
–260
(2006). http://dx.doi.org/10.1016/j.optcom.2006.07.026 OPCOB8 0030-4018 Google Scholar
A. N. Yaroslavskyet al.,
“Optical properties in selected native and coagulated human brain tissue in vitro in the visible and near infrared spectral range ,”
Phys. Med. Biol., 47
(12), 2059
–2073
(2002). http://dx.doi.org/10.1088/0031-9155/47/12/305 PHMBA7 0031-9155 Google Scholar
W. CheongS. A. PrahlA. J. Welch,
“A review of optical properties of biological tissue,”
IEEE J. Quantum Electron., 26
(12), 2166
–2185
(1990). http://dx.doi.org/10.1109/3.64354 IEJQA7 0018-9197 Google Scholar
B. R. Masterset al.,
“Mitigating thermal mechanical damage potential during two-photon dermal imaging,”
J. Biomed. Opt., 9
(6), 1265
–1270
(2004). http://dx.doi.org/10.1117/1.1806135 JBOPFO 1083-3668 Google Scholar
S. Tanget al.,
“Combined multiphoton and optical coherence microscopy,”
J. Biomed. Opt., 11
(2), 020501
(2006). http://dx.doi.org/10.1117/1.2177676 JBOPFO 1083-3668 Google Scholar
A. K. Dunnet al.,
“Influence of optical properties on two-photon fluorescence imaging in turbid samples,”
Appl. Opt., 39
(7), 1194
–1201
(2000). http://dx.doi.org/10.1364/AO.39.001194 APOPAI 0003-6935 Google Scholar
D. Kobatet al.,
“Deep tissue multiphoton microscopy using longer wavelength excitation,”
Opt. Express, 17
(16), 13354
–13364
(2009). http://dx.doi.org/10.1364/OE.17.013354 OPEXFF 1094-4087 Google Scholar
R. Cicchiet al.,
“Contrast and depth enhancement in two-photon microscopy of human skin ex vivo by use of optical clearing agents,”
Opt. Express, 13
(7), 2337
–2344
(2005). http://dx.doi.org/10.1364/OPEX.13.002337 OPEXFF 1094-4087 Google Scholar
N. JiD. E. MilkieE. Betzig,
“Adaptive optics via pupil segmentation for high- resolution imaging in biological tissues,”
Nat. Methods, 7
(2), 141
–147
(2009). http://dx.doi.org/10.1038/nmeth.1411 1548-7091 Google Scholar
B. O. WatsonV. NikolenkoR. Yuste,
“Two-photon imaging with diffractive optical elements,”
Front. Neural Circuits, 3
(6), 1
–11
(2009). http://dx.doi.org/10.3389/neuro.04.006.2009 1662-5110 Google Scholar
J. W. ChaJ. BallestaP. T. C. So,
“Shack-Hartmann wavefront-sensor-based adaptive optics system for multiphoton microscopy,”
J. Biomed. Opt., 15
(4), 046022
(2010). http://dx.doi.org/10.1117/1.3475954 JBOPFO 1083-3668 Google Scholar
C. A. Combset al.,
“Optimization of multiphoton excitation microscopy by total emission detection using a parabolic light reflector,”
J. Microsc., 228
(3), 330
–337
(2007). http://dx.doi.org/10.1111/jmi.2007.228.issue-3 JMICAR 0022-2720 Google Scholar
A. Combset al.,
“Optimizing multiphoton fluorescence microscopy light collection from living tissue by noncontact total emission detection (epiTED),”
J. Microsc., 241
(2), 153
–161
(2011). http://dx.doi.org/10.1111/jmi.2011.241.issue-2 JMICAR 0022-2720 Google Scholar
B. A. Flusberget al.,
“Fiber-optic fluorescence imaging,”
Nat. Methods, 2
(12), 941
–950
(2005). http://dx.doi.org/10.1038/nmeth820 1548-7091 Google Scholar
C. J. EngelbrechtW. GöbellF. Helmchen,
“Enhanced fluorescence signal in nonlinear microscopy through supplementary fiber-optic light collection,”
Opt. Express, 17
(8), 6421
–6435
(2009). http://dx.doi.org/10.1364/OE.17.006421 OPEXFF 1094-4087 Google Scholar
V. CrosignaniA. S. DvornikovE. Gratton,
“Enhancement of imaging depth in turbid media using a wide area detector,”
J. Biophotonics, 4
(9), 592
–599
(2011). http://dx.doi.org/10.1002/jbio.201100001 1864-063X Google Scholar
V. Crosignaniet al.,
“In vivo deep tissue fluorescence imaging of the murine small intestine and colon,”
Proc. SPIE, 8226 822609
(2012). http://dx.doi.org/10.1117/12.912841 PSISDG 0277-786X Google Scholar
T. H. ChiaM. J. Levene,
“Microprisms for in vivo multilayer cortical imaging,”
J. Neurophysiol., 102
(2), 1310
–1314
(2009). http://dx.doi.org/10.1152/jn.91208.2008 JONEA4 0022-3077 Google Scholar
F. Ayerset al.,
“Fabrication and characterization of silicone-based tissue phantoms with tunable optical properties in the visible and near infrared domain,”
Proc. SPIE, 6870 687007
(2008). http://dx.doi.org/10.1117/12.764969 PSISDG 0277-786X Google Scholar
N. Barkeret al.,
“Identification of stem cells in small intestine and colon by marker gene Lgr5,”
Nature, 449
(7165), 1003
–1007
(2007). http://dx.doi.org/10.1038/nature06196 NATUAS 0028-0836 Google Scholar
L. E. Grosberget al.,
“Spectral characterization and unmixing of intrinsic contrast in intact normal and diseased gastric tissues using hyperspectral two-photon microscopy,”
PLoS One, 6
(5), e19925
(2011). http://dx.doi.org/10.1371/journal.pone.0019925 1932-6203 Google Scholar
T. ReyaH. Clevers,
“Wnt signalling in stem cells and cancer,”
Nature, 434
(7035), 843
–850
(2005). http://dx.doi.org/10.1038/nature03319 NATUAS 0028-0836 Google Scholar
A. KlausW. Birchmeier,
“Wnt signalling and its impact on development and cancer,”
Nat. Rev. Cancer, 8
(5), 387
–398
(2008). http://dx.doi.org/10.1038/nrc2389 NRCAC4 1474-175X Google Scholar
L. G. van der FlierH. Clevers,
“Stem cells, self-renewal, and differentiation in the intestinal epithelium,”
Annu. Rev. Physiol., 71 241
–260
(2009). http://dx.doi.org/10.1146/annurev.physiol.010908.163145 ARPHAD 0066-4278 Google Scholar
N. Barkeret al.,
“Crypt stem cells as the cells-of-origin of intestinal cancer,”
Nature, 457
(7229), 608
–611
(2009). http://dx.doi.org/10.1038/nature07602 NATUAS 0028-0836 Google Scholar
M. H. RossW. Pawlina, Histology: A Text And Atlas: With Correlated Cell And Molecular Biology, Lippincott Williams & Wilkins, Baltimore, MD
(2006). Google Scholar
A. A. Heikal,
“Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies,”
Biomark. Med., 4
(2), 241
–263
(2010). http://dx.doi.org/10.2217/bmm.10.1 1752-0363 Google Scholar
|



