
|
|
1.IntroductionSince the seminal works of Folkman et al.,1,2 antivascular therapies—including vascular-targeted agents and inhibitors of angiogenesis—have emerged for destroying, modulating, and inhibiting the formation of blood vessels necessary for cancer growth. For instance, bevacizumab, a potent antiangiogenic drug, is approved for multiple cancers based on its clinical efficacy.3 Antivascular therapies increase the efficacy of radiation and chemotherapy, and these agents have become an important component of the modern armamentarium of cancer treatments.4 Targeting tumor vasculature and angiogenesis for cancer therapy has several potential advantages: (1) high tumor-selectivity since, in adulthood, normal angiogenesis is limited to wound healing and the menstrual cycle, (2) tumor vessel normalization to improve drug delivery5 (although this impairs the delivery of some agents4,6), and (3) endothelial cells do not acquire drug resistance.7 In order to fully realize these benefits, there is a critical need for developing quantitative metrics to evaluate antivascular efficacy. This concept is especially pertinent for therapeutic modalities and combination therapies that target both endothelial and cancer cells, in which differentiating antivascular activity versus direct cytotoxicity presents challenges. Like all treatment modalities, the efficacy of antivascular agents is limited by the persistence of numerous tumor escape pathways, and there is tremendous opportunity in the further development and optimization of therapeutic regimens that utilize these agents.4 However, the lack of biomarkers and screening assays of antivascular response has impeded the development of new antivascular treatments and their incorporation into combination therapies.8 Impressive advances in vascular imaging promise to elucidate new biomarkers and mechanisms of antivascular therapies. For instance, Vakoc et al. developed a rapid, noninvasive, and three-dimensional (3-D) technique—optical frequency-domain imaging—that enables label-free, longitudinal, and comprehensive volumetric imaging of tumor microvasculature up to a few millimeters deep.9 Other high-resolution imaging modalities such as fluorescence microscopy10–14 and photoacoustic tomography15 also enable resolution of individual vessels and dynamic cellular, molecular, and microstructural alterations.16 These microscopy techniques have been accompanied by development of sophisticated image analysis algorithms for 3-D microvessel tracing9,17,18 as well as morphometric and fractal analyses.9,19 In contrast, ultrasound, positron emission tomography, computed tomography, and magnetic resonance imaging (MRI) cannot resolve individual microvessels20 but, importantly, facilitate noninvasive imaging of vascular function in the clinic.16 For example, vascular MRI21 and measurements of circulating angiogenic factors have shown particular promise for monitoring “vascular normalization5” in clinical trials for new cancer treatments.22 Due to trade-offs in imaging resolution and depth, an ongoing dilemma is how best to visualize microvasculature too small to be resolved by noninvasive imaging techniques and extending to depths (greater than a few millimeters) inaccessible by high resolution optical techniques.16 In contrast to longitudinal in vivo imaging techniques, ex vivo microscopy sacrifices temporal dynamics to gain depth unlimited, high-resolution imaging at fixed time points when integrated with mechanical tissue sectioning23,24 or by methods that transform intact tissue into an optically transparent state.25 These ex vivo techniques also provide opportunities for biomarker discovery, high-throughput screening assays and help to interpret in vivo images. As an example, Inai et al. developed high-resolution electron and immunofluorescence microscopy assays to reveal that inhibitors of angiogenesis have profound impacts on microvessels within 24 h, including both shutdown and regression of existing vessels in addition to prevention of vessel sprouting and angiogenesis.11 Furthermore, “normalization5” of remaining perfused vessels was evident as a clear reduction in vessel fenestrations, tighter pericyte coverage, and reduced vessel leakiness.11 Microvessel density (MVD) quantification in “hot spots” or foci of endothelial cell proliferation, an ex vivo histology technique, was introduced by Folkman et al. over 20 years ago as a powerful prognostic indicator for many cancers,26,27 and it has since been broadly adopted as a “gold standard” metric of antivascular treatment efficacy in the clinic.28 However, Folkman et al. elegantly demonstrated that MVD misrepresents antivascular response.29 MVD can under-report antivascular efficacy because standard MVD measurements do not account for treatment-induced reductions in tumor mass.29 This decrease in tumor mass can pack the remaining microvessels together to obscure interpretation of MVD29 (Fig. 1). Therefore, MVD may falsely reject promising therapies and confound the design of antivascular treatment schemes. Moreover, characterizations of the tumor microvasculature are most commonly drawn from just a few “representative” high-magnification fields. A few early efforts have been made to automate “hot spot” identification and MVD quantification to make it more objective,30,31 but these efforts were limited to examination of a single “representative” tissue section per subject due to technical challenges in the achievement of quality staining, the lack of microscopy automation, and the availability of data storage. Therefore, the interpretation of standard MVD measurements has been further complicated by poor statistics. Due to a lack of practical alternatives, MVD continues to be the most frequently applied metric for describing antivascular effects. Fig. 1Concept of tumor total microvascularity (TMV) imaging for quantifying microvessel reduction following therapy. A theoretical schematic (not actual data), inspired by original concepts and illustrations in an elegant critical review by Hlatky et al. (Ref. 29), of tumor microvessel density (MVD) and TMV temporal profiles is shown during antivascular therapy. TMV decreases monotonically with decreasing microvessel content while MVD fluctuates due to changes in tumor volume.  With the goal of creating a broadly applicable platform that overcomes the limitations of MVD, here we introduce an efficient measurement of total microvascularity (TMV) ex vivo by integrating advances in high-throughput digital microscopy and automated analysis. Uniquely, we designed custom immunofluorescence stains and analyses to specifically identify intratumoral microvessels. We show that TMV is a superior biomarker for quantifying treatment-induced reductions in microvasculature. To address the need to reach statistical significance in data-intensive, high-resolution, and large-volume applications, we derived a mathematical model to compute the required tumor volume sampling based on empirical measurement of microvascular heterogeneity. Here, we demonstrate these concepts for TMV quantification of orthotopic pancreatic ductal adenocarcinoma (PDAC) tumors, as large as in dimension, excised from mice receiving established antivascular treatments. In summary, this platform captures the vascularity of a whole tumor using a mathematical model to set a threshold that defines the lowest limit for how much of the tumor volume needs to be sampled. This obviates the need for imaging thousands of tumor sections and, on the other hand, the risk of drawing false conclusions from an insufficient number of fields. By introducing a method for defining optimal tumor subsampling, this work offers promise for developing high-throughput assays to quantify treatment-induced microvascular alterations for therapeutic screening and development. 2.Methods2.1.Orthotopic Murine Model of PDACThis study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC). We used an orthotopic xenograft mouse model for PDAC established in our laboratory.32 For tumor implantation, a small left abdominal flank incision was made to exteriorize the pancreas of athymic Swiss male Nu/Nu mice (20 to 25 g and 6 to 8 weeks old; Cox Breeding Laboratories, Cambridge, Massachusetts). Subsequently, a suspension of 106 AsPC1 human PDAC cells in 30 μL phosphate-buffered solution (PBS, without ), containing 50% Matrigel™ Basement membrane matrix (BD Biosciences, Franklin Lakes, New Jersey) to prevent leakage of the tumor cells from the injection site was injected into the body of the pancreas. Any growth factors present in the Matrigel can potentially help “jump-start” tumor vascularization. Because Matrigel was used in all implantations, any potential angiogenic effects were captured in the untreated controls and uniformly influenced all tumors in the study. We allowed the tumor to grow for 10 days prior to the start of therapy. The terminal half-life for growth factors is typically just a few days. Hence, these components are presumably cleared and metabolized prior to day 10 when we commenced therapy. 2.2.Treatments of Orthotopic PDAC MiceTen days after the injection of AsPC1 cells, animals were randomized into the following groups: (1) no treatment; (2) a single dose of body weight cabozantinib (XL184) for antiangiogenic therapy; (3) a single dose of body weight liposomal benzoporphyrin derivative (BPD) for antivascular near infrared (NIR) photodynamic therapy (PDT; ); and, (4) a single dose of body weight BPD and body weight cabozantinib coencapsulated in a liposome for combined antiangiogenic treatment and antivascular NIR PDT. All injections were done intravenously in 200 μL sterile PBS. Mice were illuminated 90 min after BPD injection. The 690-nm light was delivered at an irradiance of . Tumor excision and TMV imaging can be applied at any time point, including within a day of antivascular agent administration, and we chose established time points used in the seminal publication demonstrating the clinical efficacy of bevacizumab ( post-treatment)33 that reflects the most common protocol for MVD quantification. Note that, here, we did not attempt to distinguish primary effects of the treatments on the microvasculature versus secondary effects resulting from inhibition of cancer cell signaling pathways and the killing of cancer cells. It is virtually impossible to do so unequivocally due to the tight interdependence of cancer cells and microvasculature,29 however, it is known that direct effects on the microvasculature are best captured at early time points.9,11 2.3.Staining Protocol for Serial Tumor SectionsAn overview of the immunofluorescence staining protocol is shown [Fig. 2(a)]. Orthotopic primary tumors were excised 2 weeks post-treatment, embedded in optimal cutting temperature compound and frozen at . A cryotome was used to cut the entire tumor into 388, 20-μm-thick cryosections. Approximately one-quarter of the sections, 105 sections (one section/70 μm), were (1) fixed in a precooled mixture of acetone:methanol for 15 min at , (2) air dried for 30 min at room temperature, and (3) washed three times, for 5 min each, in PBS with gentle agitation. A Pap Pen was used to encircle the tissue specimens for mounting small-volume blocking and staining solutions. A blocking solution (Dako Protein Block Reagent) was applied for 30 min at room temperature, followed by application of the immunostains, at monoclonal antibody (MAb) each diluted in a background reducing buffer (Dako Antibody Diluent, background reducing) for 2 h at room temperature in a humidifying chamber. Finally, the slides were washed again three times, for 5 min each, in PBS with gentle agitation, mounted (Invitrogen SlowFade Gold with 4′,6-diamidino-2-phenylindole, DAPI) with a coverslip and sealed with nail polish. Fig. 2Methods overview. (a) Work flow for tissue fixation and immunofluorescence staining. (b) Simplified image processing outline for identifying the tumor boundary and intratumoral microvessels. The microvessel content is normalized by the cell count within the tumor (using cell nuclei staining) and multiplied by the tumor volume to calculate the tumor microvessel volume () for an image mosaic of an entire tumor cross-section. The overall TMV is calculated by interpolation and summation of microvessel volume over a periodic subsampling of the entire tumor.  2.4.Confocal Fluorescence Imaging MicroscopyConfocal fluorescence imaging was performed using a commercial confocal microscope (Olympus FluoView 1000) with a 0.4 numerical aperture (NA) or a 0.75 NA objective. Excitation of DAPI, anti-human cytokeratin 8 (clone LP3K, R&D Systems, Minneapolis, Minnesota) MAb-Alexa Fluor 488 conjugates, and anti-mouse PECAM-1 (CD31; clone 390, Millipore, ) MAb-Alexa Fluor 568 conjugates was carried out using 405-, 488- and 559-nm lasers, respectively. Lasers were scanned sequentially to reduce channel crosstalk. Mosaic images of entire tumor cross-sections were collected and stitched together using the Olympus FluoView software. This method requires a manual procedure to define tumor boundaries for the mosaic. We developed a human cytoskeletal protein (cytokeratin 8) stain to demark the intracellular space of the PDAC cells, which has dual selectivity for the epithelial cancer cells because it does not react with mouse proteins and because cytokeratins are highly expressed in many epithelial cancer cells.34 CD31 (endothelial cell) and DAPI (cell nuclei) staining may be applied to quantify MVD (microvessel area is normalized by the number of cells per field). 2.5.Slide-Scanning Wide-Field Fluorescence Imaging MicroscopyIn order to efficiently image the 105 sections from an entire, orthotopic PDAC tumor, we applied a commercial slide-scanning fluorescence imaging system (Hamamatsu NanoZoomer 2.0-RS) with a 0.75 NA objective. The tumor sections were imaged in mode (). 2.6.Image AnalysesAll analyses were performed using custom MATLAB (Mathworks, Natick, Massachusetts) routines for batch processing [Fig. 2(b)]. For display and all analyses, the channels were autothresholded to reject background signal using a 10% peak intensity threshold for each channel. Wavelet transforms and image correlation coefficients among the color channels were used to eliminate bubbles and extraneous, reflective objects from the analyses. Current microvessel segmentation methods were incapable of extracting pertinent image features from image mosaics of entire tumor sections, which contain gradient backgrounds and occasional artifacts normally avoided when manually choosing individual, “representative” fields. Therefore, we developed wavelet multiresolution decomposition analyses35 for morphology-based microvessel and cancer cell segmentation. Here, MVD values (unitless) were calculated from whole slices, within viable tumor tissue only, and averaged over slices from the entire tumor rather than a more complex “hot spot” identification and calculation, which is difficult to define objectively. TMV was calculated by multiplying MVD with the viable tumor volume [Fig. 2(b)] in each slice and then summing TMV over the whole tumor by interpolation. Thus, TMV represents the total tumor endothelial cell content in units of volume, an absolute metric, rather than the fractional volume of microvessels. 2.7.Mathematical Model for Defining Tissue Volume Subsampling to Maximize Throughput Achieving Statistical SignificanceThe determination of a biomarker, such as tumor TMV, from only partial sampling of an organ or tumor leads to uncertainty in the true value. We estimated this uncertainty by using the spatial autocorrelation function of the biomarker to characterize the fluctuations between neighboring slices. These fluctuations are then used to quantify the overall noise introduced from volume subsampling. Assume an organ or tumor with equally spaced sections. is the biomarker level contained in slice and is assumed to be stationary and ergodic. We model the difference between neighboring slices as where is a Gaussian random variable of zero mean, , and is the mean of in slice space. Physically, the first term of Eq. (1) refers to the degree to which the biomarker levels (e.g., microvessels) in slice are also seen in slice . The second term reflects the biomarker levels found in slice that are not found in slice . In other words, the second term is a measure of biomarker spatial heterogeneity. Any noise due to the imaging process will also be reflected in this second term. The third term ensures the stationarity of , i.e., the collection of odd slices should have the same statistical behavior as the collection of even slices. From the autocorrelation function ,It is found that . Using the stationarity conditions and the variance (var) of the fluctuation term can be calculated. where is the standard deviation of . From this model, the standard deviation of the difference between the true biomarker level (e.g., TMV) and an experimentally measured biomarker level estimated from imaged slices can be estimated () where is the number of periodically imaged sections, is the thickness of the unimaged gap portion and is the section thickness. In this work, the tumor was well sampled to test this model of uncertainty. In practice, fewer slices with relatively large gaps between the slices can be used to fit the autocorrelation function with an exponential of unity prefactor (to force the autocorrelation to equal unity at zero lag). This fit is then used to calculate the autocorrelation for Eq. (6). Given a pilot study of organ or tumor specimens, the minimum detectable difference in the biomarker () for future studies can be estimated () where is the standard deviation of the biomarker measured across all tumors and is the number of slices per tissue specimen that would be imaged in the future study.2.8.Statistical AnalysisTreatment response data (tumor weight, MVD, and TMV) among multiple treatment groups were analyzed using a Kruskal–Wallis one-way analysis of variance (ANOVA). The Mann–Whitney test was used to report -values for specific comparisons that are not statistically significant () according to the Kruskal–Wallis ANOVA. 3.Results and Discussion3.1.Imaging MVD and Total Microvasculature in Whole TumorsWe monitored tumor microvasculature utilizing custom immunofluorescence stains and image analyses in tandem with tissue sectioning and multiarea microscopy to quantitatively image microvessels systematically throughout entire tumors [Fig. 3(a)]. The immunostains identify microvessels (endothelial cells) and epithelial cancer cells in subcellular resolution image mosaics of full tumor cross-sections [Figs. 3(b)–3(d)]. The cancer cell stain, using an anticytokeratin MAb, excludes acellular regions of necrosis and defines the boundary of viable tumor cells in serial sections to enable objective, automated segmentation of the intratumoral microvessels [Figs. 3(b), 3(e), and 4]. This identification of the tumor margin is critical as tumors are often poorly vascularized in comparison to the surrounding tissue29 [Fig. 3(e)]. In the absence of a clear visualization of the tumor margin, the dense peritumoral vasculature can confound the quantification of intratumoral microvessels. Furthermore, MVD measurements often rely on capturing changes in foci of endothelial cell proliferation, or hot spots, in a few high-magnification fields26 such that MVD statistics can suffer from interobserver variability as well as tumor heterogeneity.30 In contrast, TMV imaging estimates the total vessel volume from serial, multifield mosaics of entire tumor cross-sections for automated and objective assessments. Note that this critical identification of the tumor boundary also excludes the possibility of simply pulverizing whole tumors for biochemical assays (e.g., Western blots) because the peritumoral microvasculature obscures the quantification of intratumoral vasculature. Fig. 3Three-dimensional (3-D) tumor MVD and TMV imaging. (a) A 3-D view of select slices from a dataset of 105 PDAC tumor sections. (b) A single tumor cross-section imaged with 1.2 μm sampling illustrates selective immunostaining of mouse endothelial cells (magenta) and human epithelial cancer cells (green) to discriminate the tumor margin. Scale bar, 1 mm. (c and d) Enlarged views of the region marked in b, demonstrating visualization of (c) 4′,6-Diamidino-2-phenylindole (DAPI) staining of cell nuclei and (d) immunostaining of individual microvessels and cancer cells at the tumor margin. Scale bars, 250 μm. (e) The cancer cell immunostain defines the tumor boundary and identifies peritumoral (gray scale) versus intratumoral microvessels (magenta). Scale bar, 1 mm. 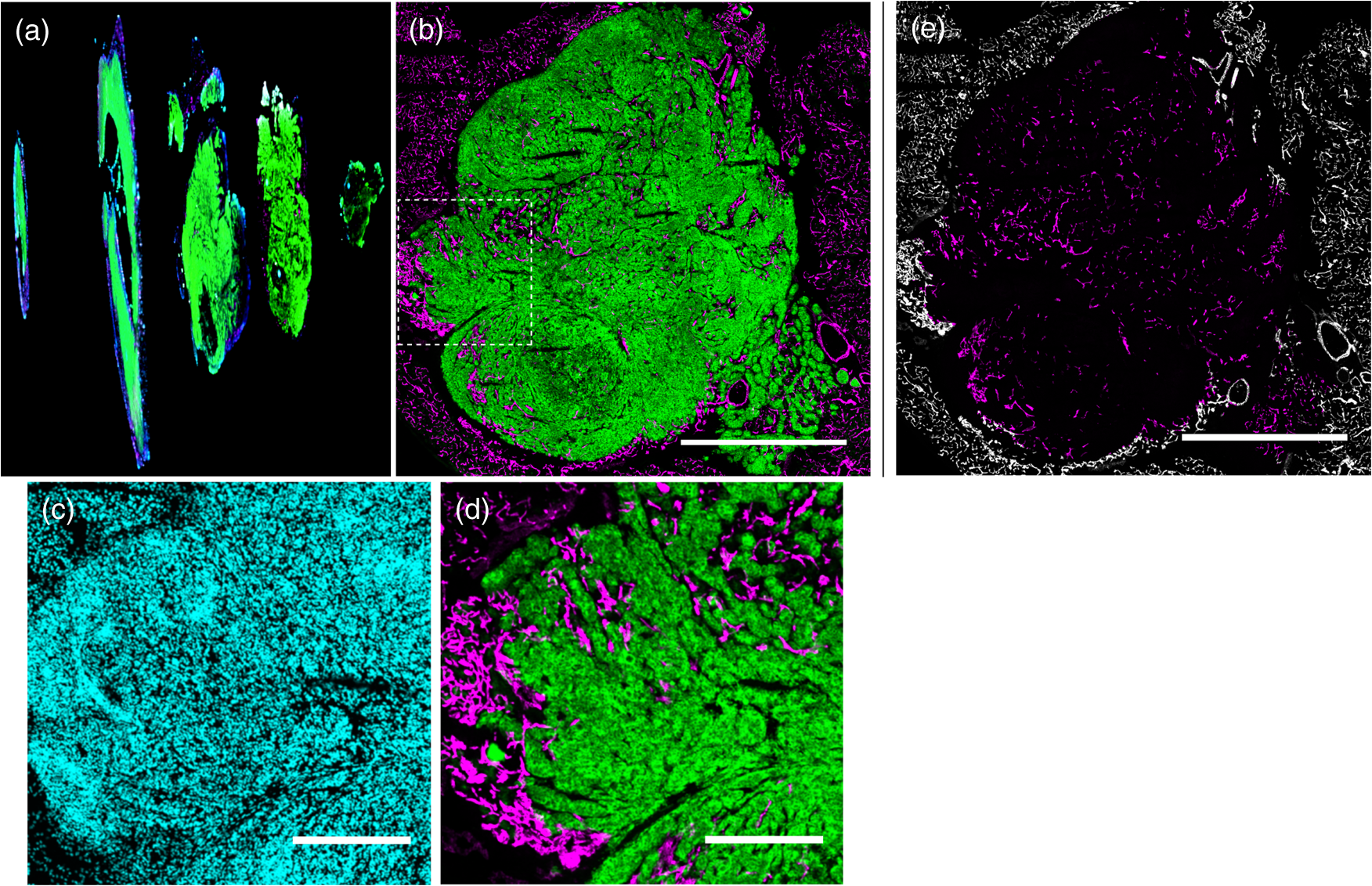 Fig. 4Wavelet-based segmentation of microvessels. Wavelet spatial frequency bandpass filtering enables automated selection of intratumoral microvessels based on morphology. This is advantageous for high-throughput extraction of immunostains from gradient backgrounds and with variable contrast over many tissue sections, as was the case here. Here, intratumoral microvessels (magenta outline) identified by software are overlaid on the raw fluorescence image of mouse endothelial cells (gray scale). (a) Selection of intratumoral microvessels using intensity thresholding fails in high-throughput applications. (b) In contrast, wavelet bandpass filtering is more robust and identifies a greater number of intratumoral microvessels.  The anti-human cytokeratin MAb for human cancer cells applied here is widely applicable to mouse xenograft models of epithelial cancers. This immunostain is selective for human cells and for cytokeratin proteins highly expressed in carcinomas, which provides a dual selectivity for clear demarcation of the tumor boundary. Prior clinical reports have demonstrated cytokeratin staining to image and classify epithelial cancers36 and micrometastases.37 Other MAbs-targeting biomarkers frequently overexpressed by cancer cells (e.g., epidermal growth factor receptors, folate receptors, and epithelial cell adhesion molecule) are also being used successfully to image and quantify tumor margins38 and ultra-rare circulating tumor cells in the clinic.39 Note that some tumor types pose difficulties for specific cancer cell staining and careful consideration will be required to design stains for other applications. 3.2.Mathematical Model for Maximizing Vascular Biomarker Quantification Throughput Achieving Statistical SignificanceFor maximal statistical power, imaging the full organ or tumor microvascular network is ideal but prohibitive. For ample, processing an entire 1-cm-diameter tissue specimen requires 170 h and 4 terabytes (TB) of data acquisition (using a Hamamatsu NanoZoomer 2.0-RS fluorescence imaging slide-scanner). However, spatial correlations of the microvasculature can greatly reduce the sampling burden. To guide efficient studies, we developed a theoretical model (Methods) to calculate the minimum number of tissue slices (tissue subsampling) needed to achieve an accurate estimate for a single organ or tumor and for an aggregate group of organs or tumors receiving a treatment regimen. The model is tunable to biological model-specific vascular heterogeneity. The appropriate subsampling can be determined from metrics of vasculature heterogeneity taken from control tissue specimens in order to balance the tradeoff between statistical confidence and experimental throughput. To validate the model in our exemplary tumor TMV biomarker imaging application, we analyzed an entire orthotopic PDAC tumor (8 mm in size) in steps of to calculate the “true” tumor TMV. We next calculated tumor TMV accuracy for varying subsamplings of the full tumor [e.g., decreasing numbers of ordered serial sections; Figs. 5(a)–5(c)] and found good agreement between empirical and theoretical confidence intervals [, tumor subsampling sizes, no fitting or free parameters; Fig. 5(c)]. The mathematical model is also applicable to models of nonmalignant disease, where the microvascular biomarker heterogeneity of organ or tumor samples can be determined and used to guide the choice of an optimal tissue subsampling frequency. Fig. 5Mathematical model for achieving maximal throughput with statistical significance. (a) Schematic of an increasing number of ordered tumor slices used to estimate tumor TMV. (b) Autocorrelation of the tumor microvasculature along the depth of an orthotopic PDAC tumor. The autocorrelation decay rate determines the optimal tumor subsampling. (c) Convergence of tumor TMV imaging accuracy as more of the tumor volume is imaged. The points are tumor TMV, confidence interval, generated by repeating the TMV calculation on permuted phase shifts of the ordered slices. Empirical confidence intervals are shown for comparison to theoretical confidence intervals generated by the mathematical model. (d) Exemplary relationship between the minimum detectable difference in tumor TMV, , and imaging an increasing number of tumors, with individual tumor samplings of 3 (0.8% of the tumor volume) or 10 (2.6% of the tumor volume) slices per tumor. 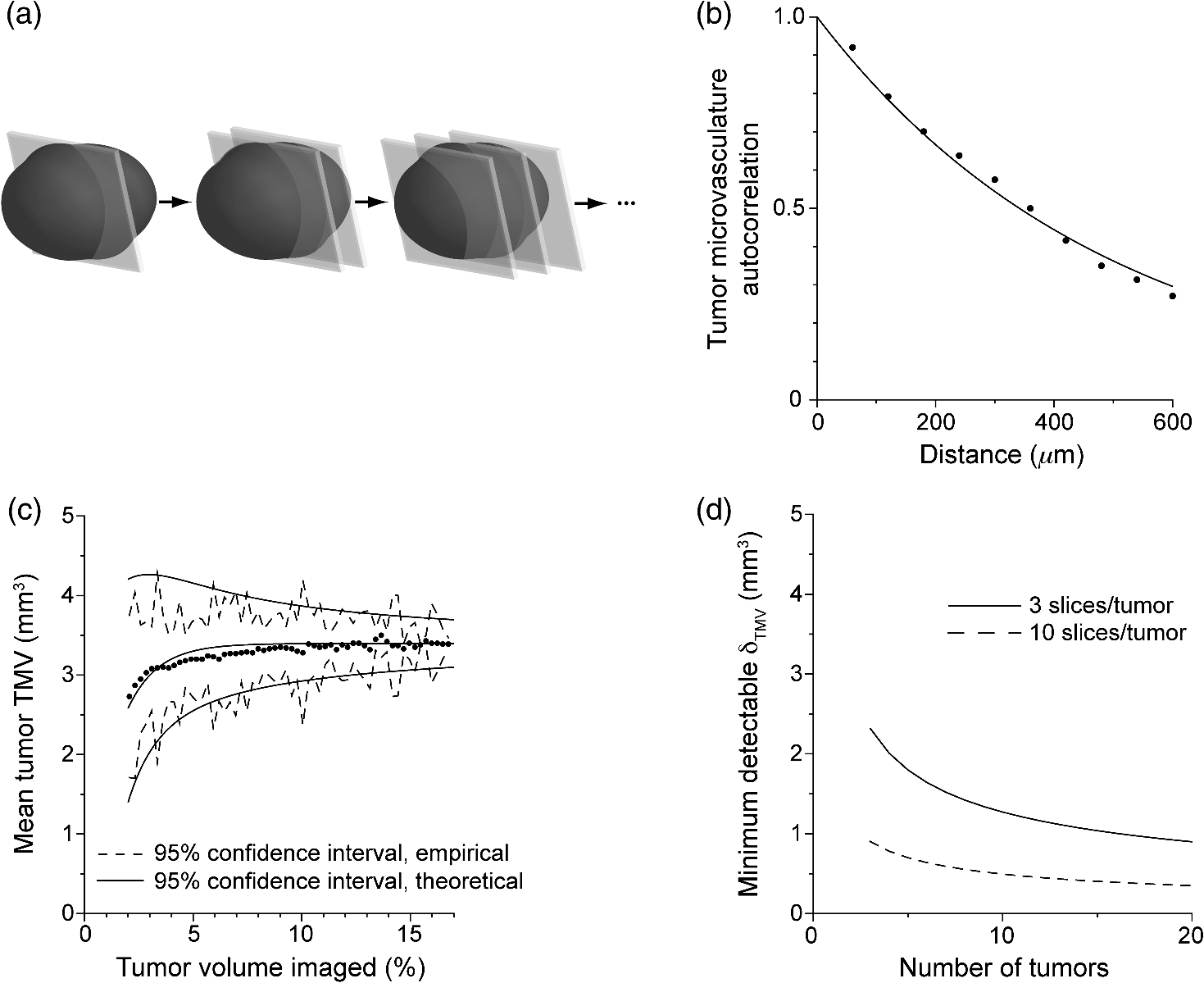 We then applied the mathematical model to determine appropriate tissue subsamplings and sample sizes (number of independent tissue specimens or tumors in this case) to resolve a desired antivascular effect size. The autocorrelation analysis characterizes vascular biomarker heterogeneity and defines the subsampling distance containing nonredundant information [Fig. 5(b)]. We define the nonredundant subsampling for a single-tissue specimen as half the decay length, , of the autocorrelation function. Here, the decay length for tumor TMV in the PDAC model is 450 μm [Fig. 5(b)], and tumor TMV accuracy reaches 85% confidence at a sampling period of 225 μm [8% of the total tumor volume, Fig. 5(c)]. Because a typical experiment includes multiple mice, it is not necessary to image at the subsampling required for a single tissue specimen; i.e., confidence improves with more specimens. As a practical guide for subsampling, a statistical calculator is provided (Appendix) that incorporates inter- and intratissue specimen vascular heterogeneity, desired effect size resolution, and sample size. The output of the calculation for the PDAC model imaged here is provided for illustrative purposes [Fig. 5(d)]. In this case, 2.6% of the PDAC tumor volume (10 slices) must be imaged per tumor for a sample size of five mice ( sections total) to achieve 95% confidence (), with a resolving power (minimum detectable difference) of a 20% change in tumor TMV. Thus investigators may plan experiments according to the desired statistical power, effect size resolution, and biomarker spatial heterogeneity. In contrast to periodic tumor subsampling, full 3-D reconstructions would require times () more labor and data storage ( sections totally). The requirement to image only a few percent of the tumor, in order to achieve sufficient statistical power to report on the whole tumor, is good news as it accelerates analysis while reducing cost. This offers promise for developing high-throughput assays. 3.3.Efficient Quantification of Antivascular Therapeutic ResponseTo demonstrate the strengths of full tumor subsampling, we performed a comparative study of MVD versus TMV in an orthotopic mouse model of PDAC subjected to different modes of treatment, (Fig. 6) resulting in either minimal or significant tumor reduction [Fig. 7(a)]. Here, MVD (dimensionless) is the mean value across the tumor depth, whereas TMV () accumulates with depth [Fig. 2(b)]. In a first experiment, we treated PDAC mice with cabozantinib (XL184), a small molecule, multireceptor tyrosine kinase (RTK) inhibitor with potent activity against MET (the receptor for hepatocyte growth factor) and vascular endothelial growth factor receptor 2 (VEGFR2), as well as other RTKs important for tumor pathobiology.40 Cabozantinib inhibits tubule formation, with no evidence of direct cytotoxicity.40 Thus, cabozantinib is classified as an antiangiogenic agent and its effects on nonvascular cells do not directly cause cell death.40 McDonald et al. have elegantly demonstrated that cabozantinib is a potent antiangiogenic agent with the added benefit of reducing a prominent escape mechanism—cancer cell migration, metastasis and growth along remaining, functional vessels as well as via lymphatic routes.4,41,42 Cabozantinib is currently in phase III clinical trials based upon its induction of antiangiogenic (through VEGFR2 inhibition) and anti-invasive (through MET inhibition) effects in preclinical models41 and in pilot clinical trials.43 Fig. 6Schematic of the treatment modalities. A simplified schematic of cell surface multireceptor tyrosine kinase (RTK) signaling triggering by growth factor (labeled ‘g’) binding and downstream, intracellular signaling cascades involving adapter proteins and other mediators (labeled with asterisks). Cabozantinib binding to RTKs leads to decreased angiogenic, proliferation, and survival signaling. Benzoporphyrin derivative (BPD)-photodynamic therapy (PDT) primarily damages mitochondria (labeled ‘m’) and its toxicity is due, in part, to subsequent apoptotic signaling cascades. 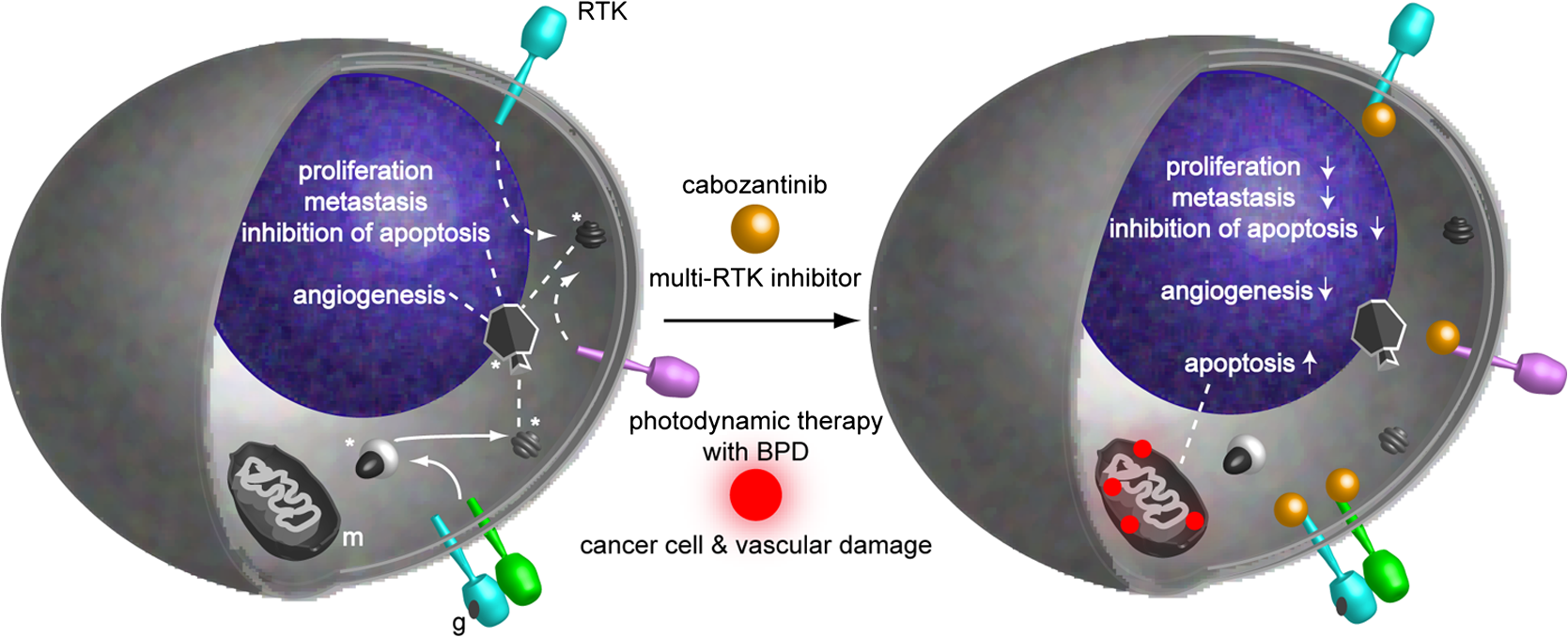 Fig. 7TMV is more sensitive than MVD imaging for reporting antivascular response. TMV imaging reports reductions in microvasculature following cabozantinib treatment, PDT, and their combination in orthotopic PDAC tumors while, in contrast, MVD imaging under-reports vascular reduction. (a) Changes in orthotopic PDAC tumor weight 2 weeks post-treatment with the monotherapies and combined PDT and cabozantinib from the data shown in (c–f) below. (b) Corresponding percent changes 2 weeks post-treatment in tumor MVD and TMV relative to no-treatment control mice. (c) The peritumoral MVD is larger than the intratumoral MVD, and the peritumoral MVD is unaffected by treatment, illustrating the importance of identifying intratumoral microvessels. (d) Intratumoral MVD shows no statistically significant changes post-treatment compared to untreated, control mice. (e) Tumor TMV is reduced for all cases and resolves statistically significant antivascular affects. (f) Significant tumor weight reduction is apparent for the treatment groups involving PDT. The results are , mice (a and f) or mice (b–e) per treatment group. Asterisks denote statistically significant differences compared to control, untreated PDAC mice, or among the indicated treatment groups, * ** *** [Kruskal–Wallis one-way analysis of variance (ANOVA) test]. In (b) and (e), note that the reason for the lower -value for the cabozantinib alone group [ mice; versus the “combo” group]—compared to the PDT alone group ( mice; versus the “combo” group)—is that the cabozantinib group has fewer mice. 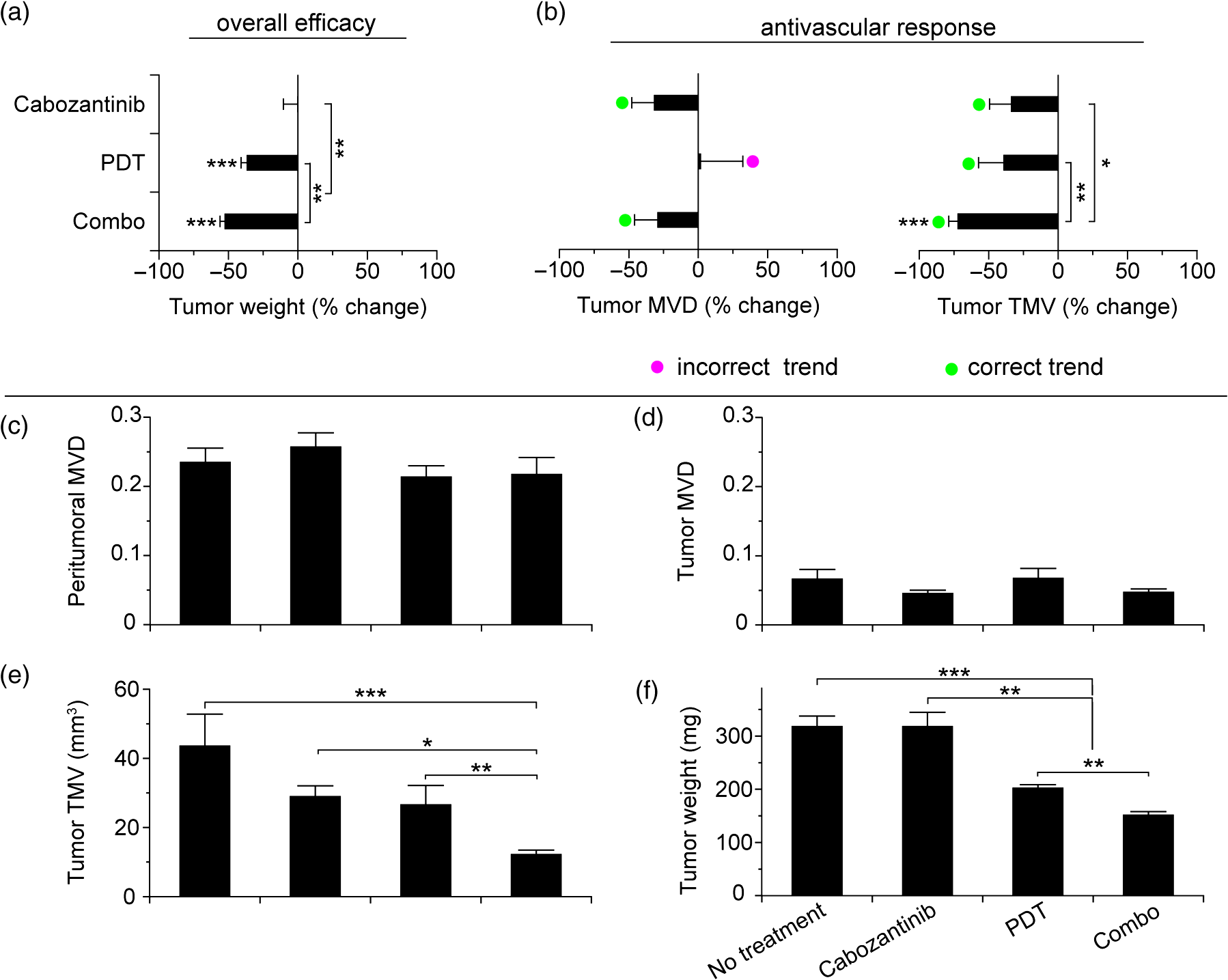 We then performed a classic antivascular treatment, PDT, on a separate group of PDAC mice using BPD activated by NIR light. PDT is a clinically utilized modality that can be utilized to directly damage vasculature. It is used routinely in the clinic for the targeted closure of neovessels in age-related macular degeneration,44,45 which is characterized by choroidal neomicrovascularization, while sparing sensitive neighboring tissues in the eye. PDT is also approved for primary and adjuvant treatment of certain cancers.45,46 Depending on the time of illumination following BPD administration, photodynamic action can selectively destroy vasculature, cancer cells or both.47–49 Here, we purposefully induced both vascular and cancer cell destruction in order to elicit a change in both overall tumor volume and microvasculature. This experimental arm tests the ability of MVD and TMV to capture antivascular response in cases where the tumor shrinks. A third group of mice received combined PDT and cabozantinib to probe for complementary effects. The single dose of cabozantinib did not affect the primary tumor burden [Fig. 7(a)], however, a trend was observed in reduced tumor MVD (, mice, 14 entire tumor cross-section image mosaics; Mann–Whitney test) and reduced tumor TMV (, mice, 14 image mosaics; Mann–Whitney test) compared to control, untreated PDAC mice [ mice, 16 image mosaics; Fig. 7(b)]. As expected, both MVD and TMV are valid metrics of antivascular activity in cases where the tumor volume is not significantly impacted. However, upon a 36% reduction in tumor weight following PDT [, mice; Kruskal–Wallis analysis of variance, ANOVA; Fig. 7(a)], tumor MVD remains unchanged while tumor TMV indicates a 39% reduction in the tumor microvasculature (, mice, 36 image mosaics; Mann–Whitney test). The final group of mice received a combination of PDT and cabozantinib, which reduced the primary tumor burden by 52% [, mice; Kruskal–Wallis ANOVA; Fig. 7(a)], an enhanced tumor reduction of 17% over PDT alone (; Kruskal–Wallis ANOVA). Both tumor MVD and TMV imaging indicated an antivascular effect [Fig. 7(b)] with a reduction in MVD (, mice, 20 image mosaics; Mann–Whitney test] and in TMV [, mice, 20 image mosaics; Kruskal–Wallis ANOVA]. However, only tumor TMV imaging was able to resolve an antivascular effect of statistical significance and also revealed a statistically significant increase in antivascular activity over PDT alone (; Kruskal–Wallis ANOVA). The data used to calculate percent changes in MVD, TMV, and tumor weight are presented in Figs. 7(c)–7(f). Collectively, TMV reported antivascular activity in all cases while MVD failed to detect reductions in the tumor vasculature when the tumor shrank in size, which tends to compact the residual microvessel content. Furthermore, TMV measurements resulted in greater statistical power than MVD measurements. These data demonstrate that TMV imaging is more suitable than MVD measurement for quantifying antivascular efficacy. 4.ConclusionsAbnormal angiogenesis and microvasculature contribute to numerous malignant, ischemic, and inflammatory disorders.50 Neovascularization is paramount to cancer growth.50,51 In addition, microvascular dysfunction and damage contribute to the etiology of rheumatoid arthritis,50 diabetes50,52,53 and Alzheimer’s disease.50,54 Furthermore, the synthesis of 3-D microvascular networks in vitro and in situ is an emerging focus of tissue bioengineering and regenerative medicine.55,56 There is a pressing need for high-throughput and high-content characterization of microvascular structural and functional properties to identify biological targets for modulating microvascular function in disease progression, therapeutic response, and tissue bioengineering. As new vascular imaging methodologies are developed to address these questions, there is a need to bridge the resolution gap between clinical and microscopic methods and to provide benchmarks for interpreting clinical images.16 Of equal importance, a further challenge is defining and achieving sufficient sampling to draw significant conclusions while maximizing throughput. A thoughtful editorial in Nature Methods calls for replacing subjective “representative” images with methods that enable reporting of quantitative, “statistically representative” image results for a true representation of the collective data.57 Until recently, the quest for quantitative microscopy has been hindered by a lack of automated microscopy systems and sufficient data storage. Bioimage informatics platforms are now emerging to facilitate extraction of objective, quantitative, and reproducible results from data-intensive, volumetric image sets.58–60 As a step towards these goals, and to realize a laboratory tool for basic biological studies and for therapeutic screening, we have integrated digital microscopy with newly developed labeling and image computation techniques to build a platform for estimating the tumor TMV. This methodology uniquely enables visualization of microvascular networks in entire organs and tumors and can be applied to benchmark noninvasive, in vivo imaging modalities. In contrast to MVD, TMV imaging is robust to changes in tumor size for evaluating antivascular treatment efficacy by quantifying changes in tumor microvasculature-associated endothelial cell volume. Therefore, TMV is superior to MVD as a metric of antivascular therapeutic efficacy and TMV imaging can be applied to screen antivascular agents and to rationally design antivascular treatment regimens. The mathematical model enables quantitative and reproducible results at the maximal throughput allowed by constraints on statistical significance informed by biomarker heterogeneity. In other words, this approach defines a statistical “speed limit” for microvascular imaging throughput based on the minimum amount of tissue volume sampling necessary to achieve statistically significant results, and the mathematical foundations are generalizable for application to a myriad of potential vascular biomarkers. These techniques remove the variability of prior subjective analyses that rely on sampling hot spots or a few random fields. TMV imaging is broadly applicable to studies of vascular disease and regenerative medicine. For instance, it can be applied to explicitly quantify perfused microvessels in tissue grafts for regenerative medicine by introducing the endothelial cell-targeted labeling probe via the circulation, in vivo, prior to the excision, preparation, and analysis of the tissue specimens.56 Here, we demonstrated the potential of TMV imaging for a critical application in cancer biology and therapy. In future studies, the platform developed here can be applied to a number of microvascular disease and basic biology studies. Metrics of vessel function (e.g., vessel permeability) and other microvascular biomarkers (such as pro- and anti-angiogenic factors) can be incorporated and the underlying concepts can be adapted for in vivo microvessel imaging.9,10,12,15 Clinical biopsies, via fine-needle aspiration, can also be analyzed by extending the mathematical model to 3-D and by designing the optimal spatial pattern for multicore sampling. For vascular biomarkers that require higher tissue samplings and 3-D tracing for morphological analyses, TMV imaging can be fully automated utilizing serial two-photon tomography, which integrates automated tissue sectioning and microscopy.24 AppendicesAppendix:Statistical Calculator SoftwareFigure 8 contains a calculator that defines the minimum amount of tissue to be imaged for accurate biomarker quantification based upon biomarker spatial heterogeneity and user input regarding statistical goals and imaging parameter preferences. The script can be copied, pasted into and run within the matlab Editor. The pilot study parameters are defined in the heading and can be adjusted interactively. AcknowledgmentsWe thank M. A. Gimbrone Jr. for a critical reading of the manuscript and insightful comments; I. Rizvi, L. A. Spring and R. P. Spring for helpful critiques; J. Zhao, J. LaGraves, E. Salomatina and P. Sherwood for expert technical assistance. This work was supported by US National Institutes of Health grants P01-CA084203 and R01-CA160998 to T. H. and F32-CA144210 to B. Q. S. ReferencesJ. Folkman,
“Tumor angiogenesis: therapeutic implications,”
N. Engl. J. Med., 285
(21), 1182
–1186
(1971). http://dx.doi.org/10.1056/NEJM197111182852108 NEJMAG 0028-4793 Google Scholar
M. A. Gimbrone Jr.et al.,
“Tumor dormancy in vivo by prevention of neovascularization,”
J. Exp. Med., 136
(2), 261
–276
(1972). http://dx.doi.org/10.1084/jem.136.2.261 JEMEAV 0022-1007 Google Scholar
H. Hurwitzet al.,
“Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer,”
N. Engl. J. Med., 350
(23), 2335
–2342
(2004). http://dx.doi.org/10.1056/NEJMoa032691 NEJMAG 0028-4793 Google Scholar
B. SenninoD. M. McDonald,
“Controlling escape from angiogenesis inhibitors,”
Nat. Rev. Cancer, 12
(10), 699
–709
(2012). http://dx.doi.org/10.1038/nrc3366 NRCAC4 1474-175X Google Scholar
R. K. Jain,
“Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy,”
Science, 307
(5706), 58
–62
(2005). http://dx.doi.org/10.1126/science.1104819 SCIEAS 0036-8075 Google Scholar
V. P. Chauhanet al.,
“Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner,”
Nat. Nanotech., 7
(6), 383
–388
(2012). http://dx.doi.org/10.1038/nnano.2012.45 1748-3387 Google Scholar
T. Boehmet al.,
“Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance,”
Nature, 390
(6658), 404
–407
(1997). http://dx.doi.org/10.1038/37126 NATUAS 0028-0836 Google Scholar
R. K. Jainet al.,
“Biomarkers of response and resistance to antiangiogenic therapy,”
Nat. Rev. Clin. Oncol., 6
(6), 327
–338
(2009). http://dx.doi.org/10.1038/nrclinonc.2009.63 1759-4774 Google Scholar
B. J. Vakocet al.,
“Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging,”
Nat. Med., 15
(10), 1219
–1223
(2009). http://dx.doi.org/10.1038/nm.1971 1078-8956 Google Scholar
E. B. Brownet al.,
“In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy,”
Nat. Med., 7
(7), 864
–868
(2001). http://dx.doi.org/10.1038/89997 1078-8956 Google Scholar
T. Inaiet al.,
“Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts,”
Am. J. Pathol., 165
(1), 35
–52
(2004). http://dx.doi.org/10.1016/S0002-9440(10)63273-7 AJPAA4 0002-9440 Google Scholar
R. P. J. Barrettoet al.,
“Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy,”
Nat. Med., 17
(2), 223
–228
(2011). http://dx.doi.org/10.1038/nm.2292 1078-8956 Google Scholar
M. E. SeamanS. M. PeirceK. Kelly,
“Rapid analysis of vessel elements (RAVE): a tool for studying physiologic, pathologic and tumor angiogenesis,”
PLoS One, 6
(6), e20807
(2011). http://dx.doi.org/10.1371/journal.pone.0020807 1932-6203 Google Scholar
N. G. Hortonet al.,
“In vivo three-photon microscopy of subcortical structures within an intact mouse brain,”
Nat. Photon, 7
(3), 205
–209
(2013). http://dx.doi.org/10.1038/nphoton.2012.336 1749-4885 Google Scholar
H. F. Zhanget al.,
“Functional photoacoustic microscopy for high-resolution and noninvasive in vivo imaging,”
Nat. Biotechnol., 24
(7), 848
–851
(2006). http://dx.doi.org/10.1038/nbt1220 NABIF9 1087-0156 Google Scholar
D. M. McDonaldP. L. Choyke,
“Imaging of angiogenesis: from microscope to clinic,”
Nat. Med., 9
(6), 713
–725
(2003). http://dx.doi.org/10.1038/nm0603-713 1078-8956 Google Scholar
M.-A. Abdul-Karimet al.,
“Automated tracing and change analysis of angiogenic vasculature from in vivo multiphoton confocal image time series,”
Microvasc. Res., 66
(2), 113
–125
(2003). http://dx.doi.org/10.1016/S0026-2862(03)00039-6 MIVRA6 0026-2862 Google Scholar
J. A. Tyrrellet al.,
“Robust 3-D modeling of vasculature imagery using superellipsoids,”
IEEE Trans. Med. Imaging, 26
(2), 223
–237
(2007). http://dx.doi.org/10.1109/TMI.2006.889722 ITMID4 0278-0062 Google Scholar
Y. Gazitet al.,
“Fractal characteristics of tumor vascular architecture during tumor growth and regression,”
Microcirculation, 4
(4), 395
–402
(1997). http://dx.doi.org/10.3109/10739689709146803 MCCRD8 0275-4177 Google Scholar
B. J. Vakocet al.,
“Cancer imaging by optical coherence tomography: preclinical progress and clinical potential,”
Nat. Rev. Cancer, 12
(5), 363
–368
(2012). http://dx.doi.org/10.1038/nrc3235 NRCAC4 1474-175X Google Scholar
J. P. B. O’Connoret al.,
“Dynamic contrast-enhanced MRI in clinical trials of antivascular therapies,”
Nat. Rev. Clin. Oncol., 9
(3), 167
–177
(2012). http://dx.doi.org/10.1038/nrclinonc.2012.2 1759-4774 Google Scholar
T. T. Batcheloret al.,
“AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients,”
Cancer Cell, 11
(1), 83
–95
(2007). http://dx.doi.org/10.1016/j.ccr.2006.11.021 CCELER 1042-2196 Google Scholar
T. Raganet al.,
“High-resolution whole organ imaging using two-photon tissue cytometry,”
J. Biomed. Opt., 12
(1), 014015
(2007). http://dx.doi.org/10.1117/1.2435626 JBOPFO 1083-3668 Google Scholar
T. Raganet al.,
“Serial two-photon tomography for automated ex vivo mouse brain imaging,”
Nat. Methods, 9
(3), 255
–258
(2012). http://dx.doi.org/10.1038/nmeth.1854 1548-7091 Google Scholar
K. Chunget al.,
“Structural, and molecular interrogation of intact biological systems,”
Nature, 497
(7449), 332
–337
(2013). http://dx.doi.org/10.1038/nature.2012.10878 NATUAS 0028-0836 Google Scholar
N. Weidneret al.,
“Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma,”
N. Engl. J. Med., 324
(1), 1
–8
(1991). http://dx.doi.org/10.1056/NEJM199101033240101 NEJMAG 0028-4793 Google Scholar
N. Weidneret al.,
“Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma,”
J. Natl. Cancer Inst., 84
(24), 1875
–1887
(1992). http://dx.doi.org/10.1093/jnci/84.24.1875 JNCIEQ 0027-8874 Google Scholar
P. B. Vermeulenet al.,
“Second International Consensus on the methodology and criteria of evaluation of angiogenesis quantification in solid human tumours,”
Eur. J. Cancer, 38
(12), 1564
–1579
(2002). http://dx.doi.org/10.1016/S0959-8049(02)00094-1 EJCAEL 0959-8049 Google Scholar
L. HlatkyP. HahnfeldtJ. Folkman,
“Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us,”
J. Natl. Cancer Inst., 94
(12), 883
–893
(2002). http://dx.doi.org/10.1093/jnci/94.12.883 JNCIEQ 0027-8874 Google Scholar
J. A. Belienet al.,
“Fully automated microvessel counting and hot spot selection by image processing of whole tumour sections in invasive breast cancer,”
J. Clin. Pathol., 52
(3), 184
–192
(1999). http://dx.doi.org/10.1136/jcp.52.3.184 JCPAAK 0021-9746 Google Scholar
K. Westeret al.,
“Automatic quantification of microvessel density in urinary bladder carcinoma,”
Br. J. Cancer, 81
(8), 1363
–1370
(1999). http://dx.doi.org/10.1038/sj.bjc.6693399 BJCAAI 0007-0920 Google Scholar
K. S. Samkoeet al.,
“Imaging tumor variation in response to photodynamic therapy in pancreatic cancer xenograft models,”
Int. J. Radiat. Oncol. Biol. Phys., 76
(1), 251
–259
(2010). http://dx.doi.org/10.1016/j.ijrobp.2009.08.041 IOBPD3 0360-3016 Google Scholar
C. G. Willettet al.,
“Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer,”
Nat. Med., 10
(2), 145
–147
(2004). http://dx.doi.org/10.1038/nm988 1078-8956 Google Scholar
O. Gireset al.,
“Cytokeratin 8 associates with the external leaflet of plasma membranes in tumour cells,”
Biochem. Biophys. Res. Commun., 328
(4), 1154
–1162
(2005). http://dx.doi.org/10.1016/j.bbrc.2005.01.074 BBRCA9 0006-291X Google Scholar
S. G. Mallat,
“A theory for multiresolution signal decomposition: the wavelet representation,”
IEEE Trans. Pattern Anal. Mach. Intell., 11
(7), 674
–693
(1989). http://dx.doi.org/10.1109/34.192463 ITPIDJ 0162-8828 Google Scholar
P. ChuE. WuL. M. Weiss,
“Cytokeratin 7 and cytokeratin 20 expression in epithelial neoplasms: a survey of 435 cases,”
Mod. Pathol., 13
(9), 962
–972
(2000). http://dx.doi.org/10.1038/modpathol.3880175 MODPEO 0893-3952 Google Scholar
S. Braunet al.,
“Cytokeratin-Positive Cells in the Bone Marrow and Survival of Patients with Stage I, II, or III Breast Cancer,”
N. Engl. J. Med., 342
(8), 525
–533
(2000). http://dx.doi.org/10.1056/NEJM200002243420801 NEJMAG 0028-4793 Google Scholar
G. M. van Damet al.,
“Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor- targeting: first in-human results,”
Nat. Med., 17
(10), 1315
–1319
(2011). http://dx.doi.org/10.1038/nm.2472 1078-8956 Google Scholar
S. Nagrathet al.,
“Isolation of rare circulating tumour cells in cancer patients by microchip technology,”
Nature, 450
(7173), 1235
–1239
(2007). http://dx.doi.org/10.1038/nature06385 NATUAS 0028-0836 Google Scholar
F. M. Yakeset al.,
“Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth,”
Mol. Cancer Ther., 10
(12), 2298
–2308
(2011). http://dx.doi.org/10.1158/1535-7163.MCT-11-0264 MCTOCF 1535-7163 Google Scholar
B. Senninoet al.,
“Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors,”
Cancer Discov., 2
(3), 270
–287
(2012). http://dx.doi.org/10.1158/2159-8290.CD-11-0240 CDAIB2 2159-8274 Google Scholar
B. Senninoet al.,
“Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice,”
Cancer Res., 73
(12), 3692
–3703
(2013). http://dx.doi.org/10.1158/0008-5472.CAN-12-2160 CNREA8 0008-5472 Google Scholar
R. Kurzrocket al.,
“Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer,”
J. Clin. Oncol., 29
(19), 2660
–2666
(2011). http://dx.doi.org/10.1200/JCO.2010.32.4145 JCONDN 0732-183X Google Scholar
A. N. Antoszyket al.,
“Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration (focus): year 2 results,”
Am. J. Ophthalmol., 145
(5), 862
–874
(2008). http://dx.doi.org/10.1016/j.ajo.2007.12.029 AJOPAA 0002-9394 Google Scholar
J. P. Celliet al.,
“Imaging and photodynamic therapy: mechanisms, monitoring, and optimization,”
Chem. Rev., 110
(5), 2795
–2838
(2010). http://dx.doi.org/10.1021/cr900300p CHREAY 0009-2665 Google Scholar
D. E. J. G. J. DolmansD. FukumuraR. K. Jain,
“Timeline: Photodynamic therapy for cancer,”
Nat. Rev. Cancer, 3
(5), 380
–387
(2003). http://dx.doi.org/10.1038/nrc1071 NRCAC4 1474-175X Google Scholar
V. H. Fingaret al.,
“Analysis of acute vascular damage after photodynamic therapy using benzoporphyrin derivative (BPD),”
Br. J. Cancer, 79
(11/12), 1702
–1708
(1999). http://dx.doi.org/10.1038/sj.bjc.6690271 BJCAAI 0007-0920 Google Scholar
K. Kurohaneet al.,
“Photodynamic therapy targeted to tumor-induced angiogenic vessels,”
Cancer Lett., 167
(1), 49
–56
(2001). http://dx.doi.org/10.1016/S0304-3835(01)00475-X CALEDQ 0304-3835 Google Scholar
B. Chenet al.,
“Combining vascular and cellular targeting regimens enhances the efficacy of photodynamic therapy,”
Int. J. Radiat. Oncol. Biol. Phys., 61
(4), 1216
–1226
(2005). http://dx.doi.org/10.1016/j.ijrobp.2004.08.006 IOBPD3 0360-3016 Google Scholar
P. Carmeliet,
“Angiogenesis in health and disease,”
Nat. Med., 9
(6), 653
–660
(2003). http://dx.doi.org/10.1038/nm0603-653 1078-8956 Google Scholar
J. Folkman,
“Angiogenesis in cancer, vascular, rheumatoid and other disease,”
Nat. Med., 1
(1), 27
–30
(1995). http://dx.doi.org/10.1038/nm0195-27 1078-8956 Google Scholar
P.-H. GroopC. ForsblomM. C. Thomas,
“Mechanisms of Disease: pathway-selective insulin resistance and microvascular complications of diabetes,”
Nat. Clin. Pract. Endocrinol. Metab., 1
(2), 100
–110
(2005). http://dx.doi.org/10.1038/ncpendmet0046 NCPEDB 1745-8366 Google Scholar
A. BrownL. R. ReynoldsD. Bruemmer,
“Intensive glycemic control and cardiovascular disease: an update,”
Nat. Rev. Cardiol., 7
(7), 369
–375
(2010). http://dx.doi.org/10.1038/nrcardio.2010.35 NRCAE6 1759-5002 Google Scholar
R. N. Kalaria,
“Neurodegenerative disease: Diabetes, microvascular pathology and Alzheimer disease,”
Nat. Rev. Neurol., 5
(6), 305
–306
(2009). http://dx.doi.org/10.1038/nrneurol.2009.72 NRNACP 1759-4758 Google Scholar
J. S. Milleret al.,
“Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues,”
Nat. Mater., 11
(9), 768
–774
(2012). http://dx.doi.org/10.1038/nmat3357 NMAACR 1476-1122 Google Scholar
L. L. Y. Chiuet al.,
“Perfusable branching microvessel bed for vascularization of engineered tissues,”
Proc. Natl. Acad. Sci., 109
(50), E3414
–E3423
(2012). http://dx.doi.org/10.1073/pnas.1210580109 PNASA6 0027-8424 Google Scholar
“The quest for quantitative microscopy,”
Nat. Methods, 9
(7), 627
(2012). http://dx.doi.org/10.1038/nmeth.2102 1548-7091 Google Scholar
J. R. Swedlowet al.,
“Informatics and quantitative analysis in biological imaging,”
Science, 300
(5616), 100
–102
(2003). http://dx.doi.org/10.1126/science.1082602 SCIEAS 0036-8075 Google Scholar
H. Peng,
“Bioimage informatics: a new area of engineering biology,”
Bioinformatics, 24
(17), 1827
–1836
(2008). http://dx.doi.org/10.1093/bioinformatics/btn346 BOINFP 1367-4803 Google Scholar
B. L. Millardet al.,
“Adaptive informatics for multifactorial and high-content biological data,”
Nat. Methods, 8
(6), 487
–493
(2011). http://dx.doi.org/10.1038/nmeth.1600 1548-7091 Google Scholar
|




