
|
|
1.IntroductionComplex I [reduced nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase] is the largest enzyme () of the mitochondrial respiratory chain and is composed of 45 different subunits. The global structure of complex I has a unique L-shaped appearance, with a 180-Å long hydrophobic sector embedded in the membrane and a hydrophilic peripheral arm protruding into the mitochondrial matrix.1 In forward mode, complex I catalyzes the transfer of two electrons from NADH, the reduced form of nicotinamide adenine dinucleotide (), to ubiquinone (Q), in a process that is coupled to the translocation of four protons to the intermembrane space.2–4 A schematic overview of the main structural domains of complex I and the specific elements involved in charge transfer to ubiquinone is presented in Fig. 1 (see also Discussion for more details). The first stage in the overall enzymatic reaction is a one-step transfer of two electrons from NADH to the prosthetic group of the complex, flavin mononucleotide (FMN), situated in the remotest hydrophilic (51 kDa) subunit, which is denoted as FMN-b.5–8 Subsequently, reduced FMN-b transfers individually the two electrons to a wire of eight consecutive iron-sulfur (Fe/S) clusters that span the extramembranous domain of the complex. The reduction of complex I-associated ubiquinone by the first electron from the terminal Fe/S cluster (N2) produces the ubisemiquinone radical (), which can then receive the second incoming electron to become fully reduced ubiquinone (ubiquinol, ). It is currently considered that Q reduction by N2 occurs in a large cleft formed by the two (49-kDa and PSST) subunits that contact the first subunit (ND1) of the membranous domain of complex I. This wide hydrophobic cavity formed at the junction between the two arms of complex I is also the place where hydrophobic inhibitors of complex I, like rotenone (ROT), or artificial electron acceptors, like menadione (vitamin ; MD), also appear to bind. Ubiquinone (coenzyme Q) is a hydrophobic molecule with an elongated structure, which can diffuse freely within the membrane. Coenzyme , the endogenous ubiquinone with ten isoprenyl subunits in its tail, is found in a large excess in intact mitochondrial innermembrane.9,10 It is likely11 that the Q-binding site in the hydrophobic cavity formed at the membrane interface between the two arms of complex I is composed of two different subsites, one for the quinone moiety near the extramembrane surface of complex I,8,11 and one for the isoprenoid chain moiety in the membrane spanning hydrophobic domain, in the ND1 subunit.11,12 Similar characteristics were reported for some complex I inhibitors as well.12,13 In addition, it has been suggested that ubiquinone can protrude deeply into the extramembrane domain of complex I by moving along an inner hydrophobic ramp.14 Consistent with this, the crystal structure of fungal complex I, which was recently resolved at high resolution in T. thermophilus,3 indicates that the ubiquinone headgroup binds close to center N2, away from the membrane surface, at the deep end of a long narrow cavity. Furthermore, it was proposed that ubiquinone can advance even further into the core of the peripheral arm of complex I, and it may thus become possible for the polar group of ubiquinone to approach and receive electrons from cluster N6a.8 Based on this idea, our current results suggest that such an event may occur with extremely low probability in ROT-bound oligomers of complex I in human leukemia Jurkat cells and is likely mediated by a conformational change induced by ROT binding to complex I oligomers. It is conceivable that human complex I arranged in dimers or higher oligomers, especially in the ROT-bound form, presents distinct features that are not displayed by the monomer of its T. thermophilus counterpart. Fig. 1Modular representation of complex I architecture. The main seven hydrophilic and seven membranous subunits, labeled according to B. Taurus Complex I subunit nomenclature,8,15 are schematically depicted. For simplicity, subunits ND3, ND6, and ND4L are comprised in a unique module. The relative positions of various subunits and Fe/S centers are only qualitatively pictured. Dashed arrows indicate possible electron transfer reactions (based on Refs. 16 and 17). The two prosthetic flavin mononucleotide (FMN) groups and their interaction with reduced nicotinamide adenine dinucleotide/reduced nicotinamide adenine dinucleotide phosphate (NADH/NADPH) are illustrated. The two rotenone (ROT) (1 and 2) and two Q ( and ) sites are specified. The dashed circle encompasses the Q-binding pocket at the interface between the hydrophobic and the hydrophilic complex I domains. Q may slide along the putative hydrophobic tunnel formed between the 49-kDa and PSST subunits, and interact with center N6a. The Nnt–complex I interaction may regulate the reduction of FMN-a via the control of the local ratio by Nnt (Ref. 8) (here, only one Nnt monomer is illustrated; the three specific domains of the monomer, I, II (extramembrane domains), and III (membrane domain), are shown).  Moreover, in a recent model derived from electron paramagnetic resonance detection of ubiquinone associated with the bovine heart complex I in situ and in proteoliposomes, it is proposed that two different species of protein-associated ubiquinone participate in the electron transport and proton pumping activity of complex I.2,15,18–20 Of these, the fast-relaxing bound ubiquinone () is close (12 Å) to and interacts with iron-sulfur cluster N2, whereas the slow-relaxing bound ubiquinone () seems to be located away () from N2, presumably in the membrane subunit ND2 of complex I.2,15,19 After receiving the first electron from N2, semireduced quinone binds with enhanced affinity to the quinone pocket and, when accepting the second electron from N2, gates the direct proton pump, which translocates two protons across the membrane.2,15 Generated ubiquinol () subsequently transfers two electrons, one by one, to the second complex I-associated quinone (). Semi- and then full reduction of causes first the uptake of two protons from the matrix side and then the dissociation of , which is released from the quinone-binding pocket to the membrane Q-pool. The latter process causes a large conformational change of the pocket that, with a piston-like movement, releases the two protons into the intermembrane space.2,15 Currently, the two pioneering complex I models by Sazanov’s and Ohnishi’s group, respectively, differ in several key aspects.3,4 For example, the x-ray structural data of fungal complex I (Ref. 3) argue against the existence of a second Q site as predicted in mammalian complex I.2,4 However, some recent independent studies appear to support Ohnishi’s hypothesis.21–23 Since the initial discovery that complex I is also capable of very slow reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidation in which a different flavin might be involved,24–26 strong evidence has been accumulated that a second FMN group (FMN-a), residing in the PSST subunit, can accept electrons from NADPH bound to a putative second dinucleotide site in the same PSST subunit.5–8 However, this reaction does not result in the overall oxidation of NADPH, so the physiological role of the second FMN is not entirely clear.8 It is, however, suggested that its main role is to control the rate of production by complex I via reduction of center N2.8 There was no indication for the second FMN site in T. thermophilus complex I.3 However, conformational changes in this large macromolecule may produce crystal disorder in the intercept region of the two arms of complex I,4 which could explain why our current data indicate the existence of a second FMN bound to complex I associated with agents like ROT, quercetin (QC), or MD, which are likely to modify complex I conformation. Moreover, T. thermophilus complex I also does not appear3 to possess the second well-documented piericidin-binding site of the mammalian complex I.8 The large body of evidences supporting the concept of two FMN and NAD(P)H sites in mammalian complex I has been thoroughly and compellingly addressed.6–8 Here we performed a detailed analysis of the properties of ultraweak photon-induced delayed photon emission [delayed luminescence (DL)] of Jurkat cells by employing treatments with ROT, as well as with MD and QC. All the three drugs share net structural similarities with ubiquinone and therefore can bind complex I inside or close to the Q-binding pocket. ROT, a familiar specific inhibitor of mitochondrial respiration,27,28 which is a steroid-type big molecule, is known to bind to two distinct, noninteracting sites on complex I.8,18,29 These ROT sites, designated ROT site 1 and ROT site 2, are most likely situated in the 49-kDa and ND1 subunits of complex I, respectively.8 ROT site 1 and ROT site 2, which display different affinities for the inhibitor,19 appear to be involved in the forward and reverse electron transfer, respectively.29 It is widely accepted that in the forward mode (site 1 involved), ROT disrupts the electron flow at the level of protein-bound ubiquinone adjacent to center N2 [i.e., (Ref. 20)], specifically by destabilizing the ubisemiquinone produced after acceptance of the first electron from center N2.20 As a direct consequence of the ROT blockage, respiration ceases and the intracellular level of NADH augments due to lack of consumption by the mitochondria. Moreover, the accumulating electrons are deviated from complex I to the surrounding molecular oxygen, thereby producing superoxide () at elevated rates, which is then released into the matrix.27 The superoxide produced by the mitochondrial electron transport chain is rapidly converted to hydrogen peroxide () by the action of mitochondrial superoxide dismutases. ROT and rotenoids, in general, have demonstrated anticancer activity, which was attributed to the induction of apoptosis.28,30 The apoptotic effects of ROT on human leukemia Jurkat T cells have been reported in several studies involving particular doses and application times.30,31 However, the reported data on ROT-induced apoptosis in Jurkat cells are extremely limited. Here, we undertook a systematic study of the ROT effects on apoptosis, NADH, and oxidized FMN () levels in Jurkat cells, under treatments of differing time and dosage. We also report, for the first time to our knowledge, the effect of ROT on clonogenic survival. The second drug used in our investigations—MD, a common stimulator of mitochondrial superoxide production, is a quinone analog also used as a chemotherapeutic agent in the treatment of leukemia.32,33 MD reduction at mitochondrial complex I,34,35 which accounts for of its metabolism,35 may result in the formation of a semiquinone with consequent superoxide production.27,35 MD was also reported to increase the activity of complex I and hence the NADH consumption by mice liver submitochondrial fractions, although it did not affect the rate of uptake in those preparations.34 In other studies, MD was found to stimulate cellular oxygen consumption.36,37 Consistent with these reports, our current results indicate that MD decreases the concentration of mitochondrial NADH in Jurkat cells by stimulating complex I activity. To further substantiate the link between DL and complex I, which was originally suggested by the partial outcomes of our ROT- and MD-based experiments, we also employed the flavonoid QC, on account of its ability to behave as a ubiquinone-like molecule.38 This phenolic compound has a quinonoid-like chemical structure, which therefore permits the binding of QC to complex I.38–40 In previous works, we determined that QC decreases the cellular content of NADH,41–43 yet the underlying mechanism is unclear, having in view the conflicting reports regarding the effect of QC on the activity of complex I. Thus, early studies reported that QC can inhibit mitochondrial respiration at the level of complex I.44 In rat brain and heart mitochondria, QC was also found to inhibit the enzymatic activity of complex I and concomitantly decrease its production of reactive oxygen species (ROS), although apparently the respiration rate was not affected.39 However, the extent of these inhibitory effects declined at increasing concentrations of ubiquinone, suggesting that QC and ubiquinone compete for binding in the specific pocket at the junction between the two arms of complex I.39 In contrast, other studies indicated that QC can bind with high affinity to complex I without inhibiting it40 and can even stimulate mitochondrial respiration.45,46 Moreover, QC appears to be able to remove the inhibition of complex I by several nonsteroidal anti-inflammatory drugs38,40 and restore the electron flow along the whole respiratory chain by acting as a coenzyme Q-mimetic molecule.38 So, it is likely that in our experiments, QC, as well as MD, stimulated the activity of complex I in Jurkat cells, hence leading to an increased rate of NADH consumption. In view of the common effects of these drugs at the level of complex I, we could investigate in more detail the connection between DL and the mitochondrial metabolism. At present, despite the fact that DL spectroscopy proved successful in some clinical applications,47–52 the origin of DL of living cells is still under debate.52,53–62 DL, also called delayed fluorescence, represents a very weak light emission elicited upon light- or UV-irradiation, which differs from ordinary (prompt) fluorescence by an unusually long decay time. Our current results clearly demonstrate that the ultraweak photon-induced delayed photon emission of Jurkat cells is quantitatively related to the mitochondrial level of NADH and that of , supporting the notion that DL is mainly produced within the mitochondrial electron transfer system at the level of complex I. The data provide novel insights into the structural and functional organization of respiratory complex I, which appears to function mainly as a dimer in its native environment. Moreover, we present evidence for an active role of secondary NAD(P)H and FMN binding sites in the functional dimer. 2.Materials and Methods2.1.Cell CulturesHuman leukemia Jurkat T cell lymphoblasts were cultured in suspension in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, penicillin, and streptomycin, at 37°C in a humidified incubator with a 5% atmosphere. Exponentially growing cells were adjusted to a density of the day before the experiment. We used hydrogen peroxide 30% solution and stock solutions of menadione sodium bisulphite dissolved in phosphate buffer saline (PBS), or dihydrated QC dissolved in dimethyl sulfoxide. Unless specified otherwise, all chemicals were from Sigma-Aldrich, (Germany). After each treatment (a list of the treatments employed in current fluorimetric and DL spectroscopy determinations is provided in Table 1), cells were washed thoroughly with PBS (pH 7.4) and resuspended in PBS at room temperature (20°C) (for DL samples, , or for spectrofluorimetry, ) or in complete medium for apoptosis assessment (). DL and fluorimetric samples were assessed immediately by DL and fluorescence spectroscopy, respectively. Cell density, viability, and morphology were examined with a CCD camera Logitech QuickCam Pro 4000 connected to an Olympus CK30 phase contrast microscope. For cell density assessment, 25 μl aliquots of the samples were diluted in PBS, stained with 0.4% trypan blue solution and to 2000 cells were imaged on a Bürker haemocytometer at the time of the DL assay. Cell count evaluation was performed both during experiments, directly by visual inspection under the microscope, and later on, by analyzing the micrographs with the use of the software ImageJ. Table 1Relative levels of reduced nicotinamide adenine dinucleotide (NADH) and oxidized flavin mononucleotide (FMNox) and the delayed luminescence (DL) yield in Jurkat cells exposed to rotenone (ROT), menadione (MD), hydrogen peroxide (H2O2), quercetin (QC), combination of QC and MD or H2O2, and antimycin A (AA).
2.2.Clonogenic Survival AssayCells were treated with ROT for 30 min at the indicated doses. Immediately after the treatment, cells were washed thoroughly with warm PBS and plated in 96-well plates at a plating density of 3, 4, or in 100 μl of complete medium per well. After three to four weeks of incubation, the plates were inspected by microscopy and the wells containing colonies with were counted. The plating efficiency was calculated by the ratio of the theoretical Poisson density of the number of negative wells observed against the initial plating density, i.e., . Clonogenic survival was calculated as the ratio between the plating efficiency of treated and control cells, respectively. 2.3.Flow CytometryCells were treated with ROT for 30 min at the indicated doses, then washed thoroughly with warm PBS, and incubated in fresh medium. After 24 h or 48 h, cells were collected, washed twice in PBS, and incubated with 5 μl Annexin fluorescein isothiocyanate (V-FITC) (Apoptosis detection kit, BD Pharmingen, California) and 2.5 μl 7-Aminoactinomycin D (7-AAD) (BD Pharmingen) in 100 μl Annexin V binding buffer for 15 min at room temperature in the dark. As recommended by the manufacturer, 400 μl of Annexin V binding buffer were then added to the samples, which were then analyzed on a Becton Dickinson FACS Calibur flow-cytometer. The excitation wavelength was 488 nm (air-cooled argon-ion laser, 15 mW) and emission was collected with 530 nm (FITC) and (7-AAD) bandpass filters. Data acquisition and analysis was performed with the use of the software CellQuest and WinMDI 2.9, respectively. Cells negative for both Annexin V-FITC and 7-AAD were considered as living cells, those positive for Annexin V-FITC and negative for 7-AAD were considered as early apoptotic cells, and those positive for both dyes were considered as late apoptotic/necrotic cells. 2.4.Delayed Luminescence SpectroscopyWe used an improved version of the ARETUSA setup,57 a highly sensitive equipment able to detect single photons. Cell samples were excited by a nitrogen laser source (Laser Photonics, Lake Mary, Florida LN 230C; wavelength 337 nm, pulse-width 5 ns, energy ). A multialkali photomultiplier tube (Hamamatsu, Iwata City, Shizuoka Prefecture, Japan R-7602-1/Q) was used as a detector for photoemission signals with wavelengths in the visible range (VIS, 400 to 800 nm), in single photon counting mode. In some determinations, two broad band ( FWHM) Lot-Oriel interferential filters, placed in a wheel between the sample and the photomultiplier, were used to select photons with wavelength of (blue) and (red), respectively, so that the experimental setup allowed one to detect only one spectral component each time. The detected signals were acquired by a multichannel Scaler (Ortec, Oak Ridge, Tennessee MCS PCI) with a minimum dwell time of 200 ns. The laser power was reduced, in some cases, to prevent the dimpling of the photomultiplier. DL measurements were done on at least three different drops from each cell sample (drop volume 15 to 25 μl) at room temperature (). PBS luminescence was subtracted from all the recordings. Photoemission was recorded between 11 μs and 100 ms after laser excitation. Raw data were accumulated in the 50,000 acquisition channels of the multichannel scaler with a dwell time of 2 μs, since at the end of this interval, the intensity of the emitted luminescence was comparable with the background value. The counts of 100 repetitions of the same run were added, with a laser repetition rate of 1 Hz. Preliminary tests showed that, under these experimental conditions, repeated runs on the same sample did not give rise to drastic monotonic changes of the intensity, whose fluctuations were inside the statistical errors. As a consequence, we excluded the possibility of photobleaching and/or photodamaging. To reduce random noise, a standard smoothing procedure63 was used by sampling the experimental points (channel values) in such a way that final data was equally spaced in a logarithmic time axis. Finally, DL intensity () was expressed as the number of photons recorded within a certain time interval divided to that time interval, to the number of living cells in the drop and to the energy of the laser. The intensity of yellow/green DL was estimated by subtracting the additive contribution of blue and red DL intensities from the VIS DL intensity. The time decay data of DL photoemission curves were fitted to an equation of the type with a variable number of exponential components. For each set of VIS, blue, green/yellow, or red DL emission data, the time decay constants () and the minimal number of exponential DL components were established from the best simultaneous fit to all DL curves obtained with control and ROT-treated cells in the respective spectral data set. Then the DL yield corresponding to each kinetic component was calculated for each individual DL curve as the product . DL decay curves exhibited, in some cases, some significant differences in emission kinetics (see Figs. 2Fig. 3Fig. 4 to 5), which reflected evident changes of the slope in three main time intervals. Accordingly, as mentioned above, it was possible to fit all DL curves corresponding to a certain treatment series with exponential components having the same time decay constants (see insets of Figs. 2 to 5) that can be clustered in time intervals differing from each other by one order of magnitude. So, to facilitate comparison between different spectral DL components, the DL yield was calculated, in some cases, in three time domains of the DL emission, corresponding to three main classes of light-emitting states—11 to 100 μs ( states), 100 μs to 1 ms (), and 1 to 10 ms (), as the integral of the -fitting function over the respective time domains. This analysis could not be performed in a consistent manner in the time domain 10 to 100 ms, as in some cases the signal-to-noise ratio was too low within this region. Fig. 2Kinetics of delayed photoemission in VIS by Jurkat cells exposed for 30 min to dimethyl sulfoxide (DMSO) (control) or to the indicated doses of ROT. The time elapsed after UV-excitation of the cell sample is presented on the abscissa. Each curve was obtained by fitting the data to the equation . Insets present the ROT-dose dependence of the delayed luminescence (DL) yield (i.e., the product ) corresponding to each kinetic component. The characteristic times () of individual exponential components are indicated. The first component () is not specified and was disregarded in further analysis.  Fig. 3Kinetics of delayed photoemission in red by Jurkat cells exposed for 30 min to DMSO (control) or to the indicated doses of ROT. Each curve was obtained by fitting the data to the equation . Insets, the ROT-dose dependence of the DL yield (i.e., ) produced by three dominant classes of kinetic components. The characteristic times () of individual exponential components are indicated. The first component () is not specified and was disregarded in further analysis. 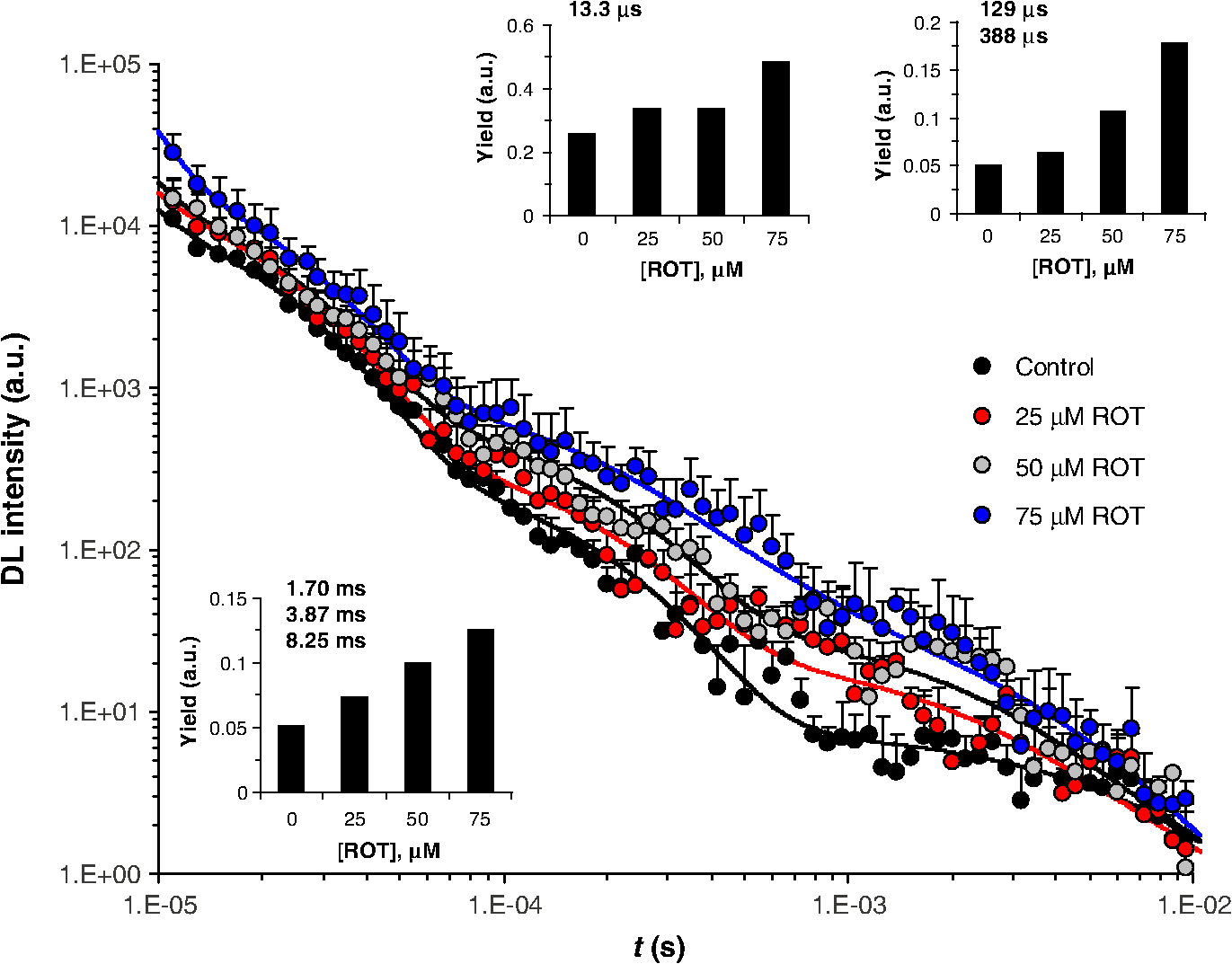 Fig. 4Kinetics of delayed photoemission in VIS by Jurkat cells exposed for indicated periods to DMSO (control) or to 50 μM ROT. Each curve was obtained by fitting the data as in Fig. 2, using the characteristic decay times indicated therein. Insets, the DL yield dependence on treatment duration. Other details are as in Fig. 2.  Fig. 5Kinetics of delayed photoemission in red by Jurkat cells exposed for indicated periods to DMSO (control) or to 50 μM ROT. Each curve was obtained by fitting the data as in Fig. 3, using the characteristic decay times indicated therein. Insets, the DL yield dependence on treatment duration. Other details are as in Fig. 3. 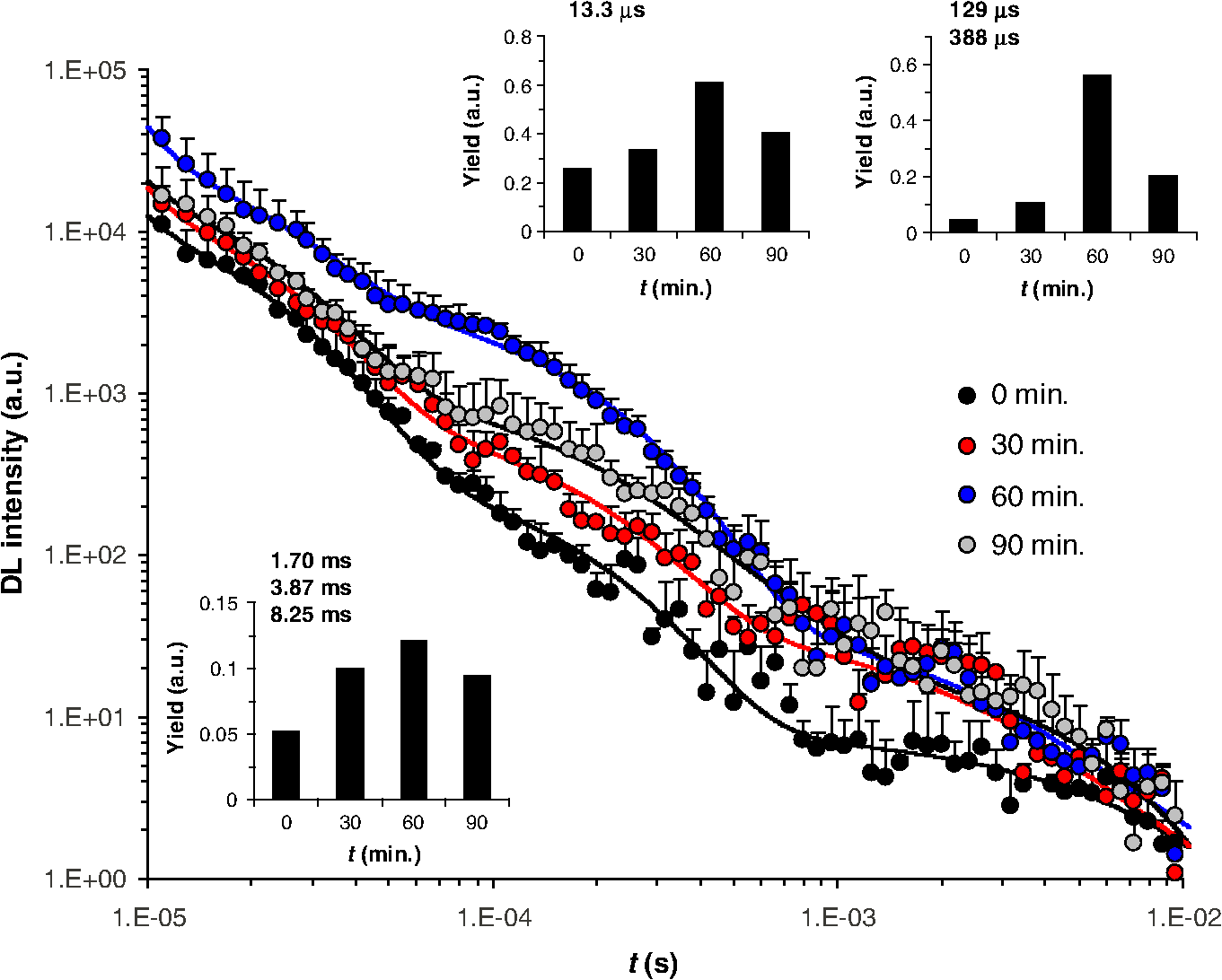 2.5.SpectrofluorimetryFor the determination of NADH and levels, 2 ml cell samples prepared in PBS as described above were placed into a covered quartz cuvette maintained at 20°C under continuous stirring in a Horiba Jobin Yvon, (New Jersey) spectrofluorimeter equipped with two monochromators. All slits were set to 5 nm. After 5 min allowed for thermal equilibration, emission spectra were collected in triplicate, with excitation at 340 nm for NADH64,65 and at 450 nm for .66 Fluorescence intensity was averaged, corrected for background, and then normalized to cellular density. The mitochondrial level of NADH, , was quantified by the ratio of fluorescence emitted at 450 nm to that emitted at 374 nm and is considered to be not affected by inherent artifacts related to cell movements, sample inhomogeneities, etc.67,68 In control (nontreated) cells, this ratio was (). The NADH fluorescence ratio obtained with treated cells was normalized to the corresponding mean value of the control cells (1.86). As discussed in more detail in the paper, our results are consistent with the notion that mitochondrial NADH constitutes the major component of cellular NADH under our conditions, in agreement with other reports [Refs. 65 and 67 to 69; see also Ref. 64 for a review]. It is also assumed that the concentration of NADPH, whose photophysical properties are virtually identical to those of NADH, is much lower than that of NADH.64,70 This idea is also strongly supported by the strong correlation found in the present study between the fluorescence of NADH and that of FMN in cells treated with complex I-targeting agents (discussed below). The level of is expressed as the fluorescence emission at 515 nm with excitation at 450 nm.65,66 In QC-based experiments, due to consistent spectral overlap between the two fluorescent species, FMN fluorescence was recorded with excitation/emission at , where QC fluorescence is very weak.41,43 This assessment may somewhat overestimate emission from FMN due to contribution from QC. Nevertheless, this further strengthens the current conclusion that QC decreases the level of . As discussed below, the results obtained in this study suggest that the fluorescence we detected at 515 nm originates mainly from FMN rather than flavin adenine dinucleotide (FAD) or other mitochondrial flavins, which, due to their similar optical properties, are virtually indistinguishable by spectrofluorimetric means. In kinetic fluorimetric recordings, cell suspensions prepared in PBS at a density of were measured under continuous stirring at 37°C. Fluorescence was recorded in kinetic mode, with alternate excitation at 340 and 450 nm, respectively. Excitation pulses were repeated every 22 s. Emission detected at . 340 nm (NADH) and at . 450 nm (FMN) was corrected for background and normalized to cellular density. At indicated time points, the drugs were added directly to the cuvette, without interrupting the recording. 3.Results3.1.Effect of ROT on Clonogenic Survival and ApoptosisROT decreased the clonogenic survival of Jurkat cells in a dose-dependent manner [Fig. 6(a)]. In acute (30 min) exposure to ROT, the corresponding median effect concentration (required to reduce clonogenicity to 50%) and Hill-type coefficient of ROT were estimated to be and , respectively. As also discussed below, these results indicate that the cooperative interaction between at least two ROT-binding sites contributes to the biological endpoint (cell death). Fig. 6Dependence of clonogenic survival (a) and apoptosis (b) on the dose of rotenone. Jurkat cells were exposed for 30 min to the indicated doses of rotenone and then processed for assessment of clonogenic survival and early/late stages of apoptosis as described in Materials and Methods. Data in (a) were fitted to the equation with and . In (b), early and late apoptotic cell fractions were evaluated 24 and 48 h after drug removal.  ROT also induced apoptosis dose-dependently, which correlated well with the clonogenic survival [Fig. 6(b)]. The kinetics of apoptosis induction appeared to be relatively slow, since 48 h after the treatment, a significant number of cells were found in early apoptotic phases. The specific evolution of apoptosis through early/late stages strongly indicated that ROT preferentially induced apoptosis, not necrosis in Jurkat cells. This conclusion was further substantiated by forward and side scatter analysis (not shown). 3.2.Effect of ROT, QC, MD, and on NADH and LevelsWe investigated by spectrofluorimetric means the effect of ROT on the mitochondrial level of NADH and . As expected, steady-state measurements (Fig. 7) and kinetic recordings (not shown) showed that in ROT-treated Jurkat cells, the NADH level increased substantially. Moreover, inhibition of complex I by ROT was additionally confirmed by separate fluorimetric determinations of the level of mitochondrial superoxide, which displayed a consistent increase after addition of ROT (not shown). Interestingly, contrary to our expectations based on a leading effect of the cellular redox state (see, for example, Ref. 65), the level of also increased after addition of ROT (Fig. 7), probably reflecting a direct effect of NADH on the rate of FMN oxidation at the level of complex I in this cell type (discussed below). From steady-state measurements carried out after treatments with increasing doses of ROT [Fig. 7(a)], the microscopic dissociation constant associated with ROT binding to complex I was estimated to be , and the Hill coefficient of 2.0 to 2.4 must indicate cooperative binding of at least two molecules of ROT to a complex I oligomer, since the Hill coefficient for ROT binding in complex I in the forward mode (site 1 involved) was determined to be equal to 1.18 Importantly, our data, corroborated by additional results presented below, suggest that in intact Jurkat cells, complex I functions mainly as a dimer in which two molecules of ROT bind cooperatively to the corresponding ROT sites 1. Fig. 7NADH and FMN autofluorescence depends on the mitochondrial metabolic state. [(a) and (b)] Effects of 30 min rotenone-treatment on NADH and levels in Jurkat cells. (a) The relative ligand concentration (normalized to control) depended in a Hill-like fashion on the dose of rotenone. The data were fitted to the equation: . For the NADH data set, , , and . For the data set, , , and . (b) Dependence of NADH and levels on the duration of rotenone exposure, for a fixed dose of 50 μM rotenone. (c) NADH and concentrations after rotenone (ROT), menadione (MD), quercetin (QC), and treatments are strongly correlated. The line () was obtained by linear fit to the data (circles). Effects of ROT, MD, QC, and applied alone or in combination (circles) are compared to the effect of antimycin A (AA, square). All data and the corresponding treatments are indicated in Table 1. Kinetic traces of NADH (d) and FMN (e) autofluorescence in stirred suspensions of Jurkat cells on 37°C exposure to ROT and 2,4-dinitrophenol (DNP). At indicated time points (arrows), drugs were added in the sequence: three steps of 50 μM ROT each, then 75 μM DNP, and finally 2.5 mM HCl and 5 mM NaOH. The final pH was 6.1 and 7.9 after addition of HCl and NaOH, respectively. Kinetic traces of NADH (f) and FMN (g) autofluorescence in stirred suspensions of Jurkat cells on 37°C exposure to ROT, AA, and DNP. At indicated time points (arrows), drugs were added in the sequence: 50 μM ROT, 25 μM ROT, 4 μM AA, 4 μM AA, 75 μM DNP, 1 mM HCl, and 2 mM NaOH. The final pH was 6.9 and 7.6 after addition of HCl and NaOH, respectively. Traces in d-g are representative for three different experiments.  The data presented in Fig. 7(b) indicate that the effect of ROT was complete in . Indeed, treatments with 50 μM ROT for 30, 60, or 90 min had similar effects on NADH and levels. However, the slight reduction of , which was visible after the 90-min treatment, corroborated by results from DL spectroscopy (discussed below), indicate that a conformational change in complex I may be induced after longer treatments with ROT. We also investigated the effect of QC, MD, and on the mitochondrial concentration of NADH or . In agreement with our previous reports,41–43 current measurements confirmed that QC decreases the NADH level in Jurkat cells (this point will be discussed in more detail in the next sections). In addition, QC also depressed the level of (Table 1). MD had similar effects, considerably decreasing the concentration of both NADH and in Jurkat cells (Table 1). It should be noted that the former effect is consistent with previous findings that MD increases the NADH consumption by mice liver submitochondrial fractions.34 High concentrations of (100 and 500 μM) applied for 20 min (which were found to induce substantial apoptosis in Jurkat cells42,62) did not cause significant variations in and produced only a slight decrease in [] (Table 1). Taken together, the steady-state fluorimetric data presented here demonstrate a strong linear correlation between the level of NADH and that of in Jurkat cells treated with ROT, MD, QC, combination of QC and MD, and [Fig. 7(c), Table 1]. The Pearson correlation coefficient associated with these data was . A similar atypical behavior of flavin fluorescence was previously reported in muscle tissue71 and human monocytes,72 where ROT was found to increase flavoprotein emission, but the reason for this was unclear. In addition, we found that under similar conditions, antimycin A (AA), an inhibitor of respiratory complex III, induced a substantial increase of but had no measurable effect on [] under the current conditions [Fig. 7(c)]. A number of studies have identified -lipoamide dehydrogenase (LipDH), FAD bound in the electron transfer protein (ETF), free oxidized FAD/FMN, enzymes of fatty acid oxidation, or lipofuscin as possible contributors to flavin autofluorescence;73–76 for a comprehensive recent review of possible cellular sources for autofluorescence and their relation to the metabolic state, see Ref. 77. Our data strongly suggest that the main flavoprotein detected in Jurkat cells under our conditions is most likely complex I-related FMN. It should be mentioned that in our determinations, FMN fluorescence was relatively weak (10 times lower than NADH fluorescence). It is generally recognized that the fluorescence of flavins can be substantially quenched (20- to 100-fold) upon binding to proteins that are involved in electron transfer.78 However, the FMN fluorescence of complex I is measurable.79,80 In addition, if FAD were the major component of the flavin autofluorescence detected in our assay, we would have observed a significant decrease in flavin fluorescence in both ROT- and AA-treated cells.65,81,82 The same also stands for lipoamide dehydrogenase, another major flavoprotein that could contribute to the observed fluorescence signal. Since the redox state of its flavin cofactor is in tight equilibrium with the mitochondrial pool,65 autofluorescence originating from LipDH should be inversely related to the mitochondrial metabolic activity, and hence to NADH autofluorescence. However, this was not the case here. Moreover, the changes in cellular ROS levels were not correlated with the variations in the flavin fluorescence observed in our assay. Indeed, using a general oxidative stress indicator as well as a mitochondrial superoxide indicator, we found that, in treatments similar to those employed in this study, both ROT and MD induced a substantial increase, whereas QC induced a substantial decrease of the ROS production (manuscript in preparation). In addition, the accumulation of lipofuscin pigments, which can generate significant autofluorescence, was reported to be induced by ROT (Ref. 83) as well as by MD (Ref. 84) via oxidative stress. However, under our conditions, the effects of ROT and MD on flavin autofluorescence were opposite, hence arguing against the idea that our recordings reflect fluorescence changes originating from lipofuscin. Furthermore, the parallel variations in both NADH and flavin autofluorescence manifested immediately after addition of ROT [Figs. 7(d) and 7(e)] or MD (not shown), whereas augmentation of lipofuscin fluorescence is expected to develop on a much longer time scale after exposure to ROT.83 In certain systems, a significant fraction of flavin-associated autofluorescence can be provided by the FAD prosthetic group of the ETF.75 However, since ETF’s fluorescence is not NADH-dependent and, moreover, is substantially quenched upon ETF reduction treatments with complex III inhibitors,75 our data clearly indicate that the major flavin detected in our measurements is not related to ETF. Possible variations in intracellular pH are also unlikely to be the underlying cause of the atypical flavin response observed in our assay. To induce variations in intracellular pH, we used the uncoupler of oxidative phosphorylation, 2,4-dinitrophenol (DNP), which can reduce the pH gradient across cellular membranes by acting as a protonophore. First, sequential addition of the mitochondrial inhibitors ROT or AA to Jurkat cell suspensions produced a prompt and substantial stepwise augmentation of NADH fluorescence [Figs. 7(d) and 7(f)], whereas flavin autofluorescence was apparently responsive to ROT, but not to AA at effective concentrations () [Figs. 7(e) and 7(g) and data not shown]. These levels of inhibition produced increases in NADH and flavin fluorescence that were similar to those obtained in steady-state measurements described above [Fig. 7(c)]. Since the simultaneous block of complexes I and III should produce high levels of reduced flavins downstream of complex I and upstream of complex III, the ROT-AA combination should produce a large decrease in flavin autofluorescence if complex II- or ETF-associated FAD were the main fluorescent flavin in Jurkat cells, which was not the case here. Even more, high concentrations of AA (150 μM) added after exposure to 50 μM ROT induced a twofold increase in flavin fluorescence (not shown), suggesting that the actual increase in FMN fluorescence is probably higher if we compensate for the contribution of the presumably decreased FAD fluorescence to the detected signal. After addition of 75 μM DNP, a rapid and consistent decrease in both NADH and flavin autofluorescence was observed [Figs. 7(d) to 7(g)]. This strong NADH response to DNP was unlikely to be the effect of a pH change, since subsequent addition of HCl or NaOH affected NADH autofluorescence to a modest extent in comparison with the wide variation elicited by DNP. In the presence of ROT and DNP, flavin autofluorescence was unaffected by the addition of 2.5 mM HCl, but decreased after subsequent addition of 5 mM NaOH [Fig. 7(e)]. In the presence of ROT, AA, and DNP, flavin autofluorescence responded with a twofold increase on addition of 1 mM HCl and then decreased three times upon further addition of 2 mM NaOH [Fig. 7(g)]. NADH and flavin fluorescence emissions were strongly correlated during exposure to ROT [Pearson coefficient for data in Figs. 7(d) to 7(e) and in Figs. 7(f) to 7(g)] and after addition of DNP [ in Figs. 7(d) to 7(e) and in Figs. 7(f) to 7(g)], irrespective of the induced pH variations. The typically expected effect of DNP is to decrease NADH fluorescence and increase flavin fluorescence by inducing an oxidized state of the mitochondria.85 DNP at higher concentration (250 μM) was reported to induce a relatively small decrease (0.1 pH units) in the intracellular pH, while evoking substantial plasma-membrane and mitochondrial depolarization.86 We also found that under our conditions, all the three agents, ROT, QC, and MD, produced substantial mitochondrial depolarization (not shown), indicating that the currently observed NADH-flavin positive correlation is not due to variations in the mitochondrial transmembrane potential. MD- or QC-based determinations also confirmed the parallelism of the NADH and flavin fluorescent signals under conditions where autofluorescence was depressed after addition of the oxidative stressor (not shown). Taken together, all these results and observations strongly support the notion that in our assay both NADH and flavin fluorescent signals arise primarily from the mitochondria, since they display strong responses to various mitochondrial inhibitors, and also indicate that the major fluorescent flavin detected in our assay is FMN. Some relevant molecular aspects will be further discussed in Sec. 5. As analyzed in more detail therein, the most likely interpretation that can explain all of the outcomes in the present study is that ROT, QC, and MD induce a complex I conformational change in which the flavin radical produced after reduction of center N3a by dislocates within the FMN binding cavity of complex I and reacts with molecular oxygen. Regenerated then rebinds with high affinity to its site. Since the dissociation of from the FMN site of complex I is stimulated by NADH,87 increased levels of NADH would lead to more FMN released within the binding cavity of complex I and thus to an apparent prolongation of the FMN dwelling time in the oxidized state before rebinding to the FMN site. In addition, unbound FMN is much more fluorescent than in its bound form and has a quantum yield that is 10 times higher than that of FAD,88 hence explaining the increased fluorescence of flavin at increased levels of NADH. 3.3.Effect of ROT on Delayed LuminescenceTo probe the relationship between the ultraweak photon-induced delayed photoemission and complex I, we investigated the effect of ROT, QC, MD, and on DL. DL spectroscopy in VIS revealed a notable enhancement of DL following ROT treatments of Jurkat cells. Light emission following UV excitation presented a complex kinetic profile, which was well fitted by a linear combination of four exponentials, with characteristic decay times of 22.9 μs, 140 μs, 1.40 ms, and 8.50 ms, respectively (Fig. 2), indicating the existence of four main classes of light-emitting states. With treatments of 30 min duration, the corresponding yields associated with the total number of excited states of each component increased with the dose of ROT (Fig. 2, insets). Under the same treatment conditions, the kinetics of delayed red-light emission exhibited six dominant exponential components, with characteristic decay times of 13.3 μs, 129 μs, 388 μs, 1.70 ms, 3.87 ms, and 8.25 ms, respectively (Fig. 3). Some of these components provided an extremely low DL yield in certain treatments. Therefore, to obtain a unitary description and gain relevant insights into the effect of ROT on red DL, we collected the contributions of some adjacent DL components as shown in the insets of Fig. 3. Together, these data show that the overall effect of ROT was to stimulate red DL. Next, we maintained the ROT dose constant and varied the treatment duration from 30 to 60 and 90 min, respectively. Delayed luminescence in VIS (Fig. 4) as well as in the red region of the spectrum (Fig. 5) presented a biphasic dependence on the treatment time, exhibiting a maximum in 60-min treatments. In particular, a marked () increase was observed in the delayed red-light emission by a distinctive component with decay time constant of 129 μs. We have so far determined the yields and time decay constants of the dominant VIS and red DL components. After analyzing in the same manner, the delayed light emission in the blue and the green/yellow regions of the spectrum, which displayed similar features with regard to the dependency on the ROT dose and treatment duration, we could notice that some kinetic components were not common to all spectral components of DL. Therefore, to obtain a more general description of the DL characteristics and perform a consistent comparison between different spectral domains, we considered three distinct classes of light-emitting states, namely one class of fast-decay states (, lifetime ) and two classes of intermediate- (, lifetime ) and slow-decay states (, lifetime ); this approach, together with the individual decay times determined above, proved particularly useful in identifying the specific electron transfer rates in complex I (see Discussion for more details). We should stress that, taking into consideration the specific DL decay times obtained above for the VIS and red spectral regions (Figs. 2 to 5), this actual classification provided the most consistent analysis of the data and, moreover, allowed us to describe the green/yellow DL emission in terms of emission kinetics with similar dominant decay times. The results collected in Fig. 8 show that the fast-decay () states emitted predominantly blue light, with somewhat lower contributions from red- and green/yellow-light emitting states, whereas states and appeared to emit mostly in the green/yellow region, with some significant blue- and red-light emission from recorded only after ROT treatments of 60 min. Fig. 8Spectral analysis of the DL yield as a function of (a) the ROT dose in Jurkat cells exposed for 30 min to the indicated concentrations of ROT or (b) the ROT treatment duration, at a fixed dose of 50 μM ROT. Blue, green/yellow, and red DL yields were computed for three different classes of states, , , and , which emit on different timescales (see text for further details). 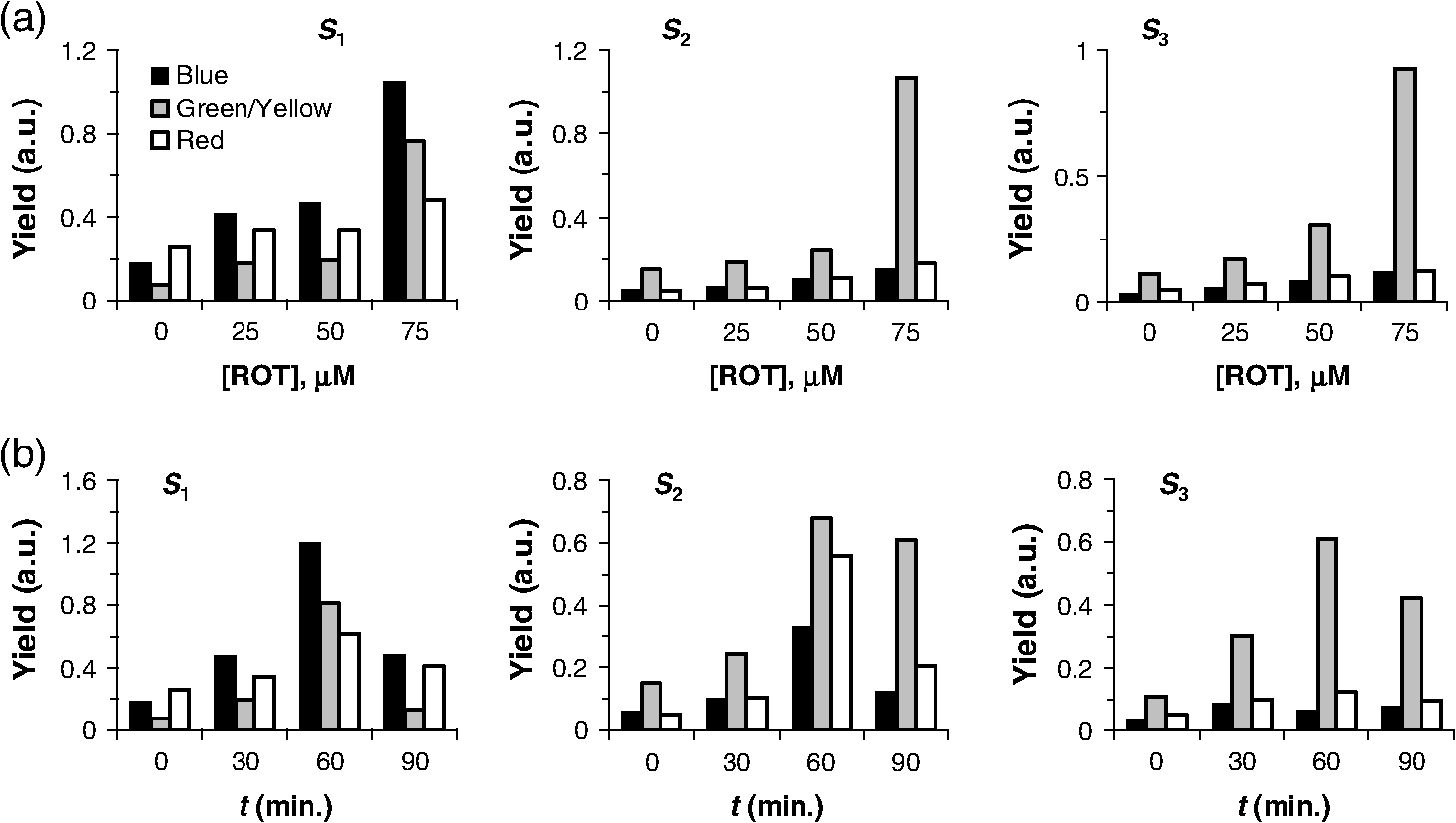 The data presented in Fig. 7(b) indicate that under our conditions, the effect of ROT on both NADH and levels was complete in . The slight attenuation of the effect that was observable after longer treatments (90 min) was reflected more prominently by the DL yield (Figs. 4 to 8). It is conceivable that mitochondrial complex I may become insensitive to ROT after long exposure to the inhibitor via a slow deactivated/activated state transition,89 which could be regulated metabolically in intact cells. Hence, this mechanism would explain the apparent recovery from inhibition, which, under our conditions, seems to manifest in exposures of increased duration. 3.4.Correlation Between DL and the Level of NADH andDL spectroscopy and NADH/ fluorimetric data were collected after treatments with ROT, QC, MD, and , or QC in combination with MD or . Consistent with our previous reports,42,62 QC and MD significantly inhibited the ultraweak photon-induced delayed photoemission of Jurkat cells in a dose- and time-dependent manner (Table 1). elicited as well an inhibitory effect on DL, but to a much lesser extent (Table 1). Combination of QC and MD at high dose also consistently decreased the VIS-DL yield, whereas combination of QC with a high dose of exerted a more reduced effect (Table 1). Taken together, the data revealed a strong correlation between the total DL yield in VIS and the mitochondrial concentration of NADH or [Fig. 9(a)], which also manifested robustly for all classes of states , , and (not shown). The dependence of the total VIS-DL yield on both and [] was very well fitted by a modified Hill equation [Fig. 9(a)]. Interestingly, the apparent Hill coefficient of both ligands was estimated to be 4.3 to 4.4, which is approximately two times higher than the value estimated above for the binding of ROT to oligomeric complex I. Fig. 9The dependence of the VIS (a), blue (b), green/yellow (c), and red (d) total DL yield on the relative concentration of NADH or , obtained after treatments of Jurkat cells with ROT, MD, QC, or . The total DL yield was calculated from emission over the time interval of 11 μs to 10 ms. The data (collected in Table 1) were fitted to the equation (curves), where is the relative ligand concentration. The fit to VIS-DL data was done with set to 21.0, the maximal value obtained for the first exponential component of the photoemission curves in Figs. 2 and 3. The fit to spectral components was done with fixed and , which were considered to be a certain fraction of the corresponding VIS values. The blue and red fractions were set to 0.291 and 0.318, respectively, which were equal to the maximal values observed from all measurements. The remaining fraction, 0.391, was attributed to green/yellow DL. Parameters derived from the fit are presented in Table 2. 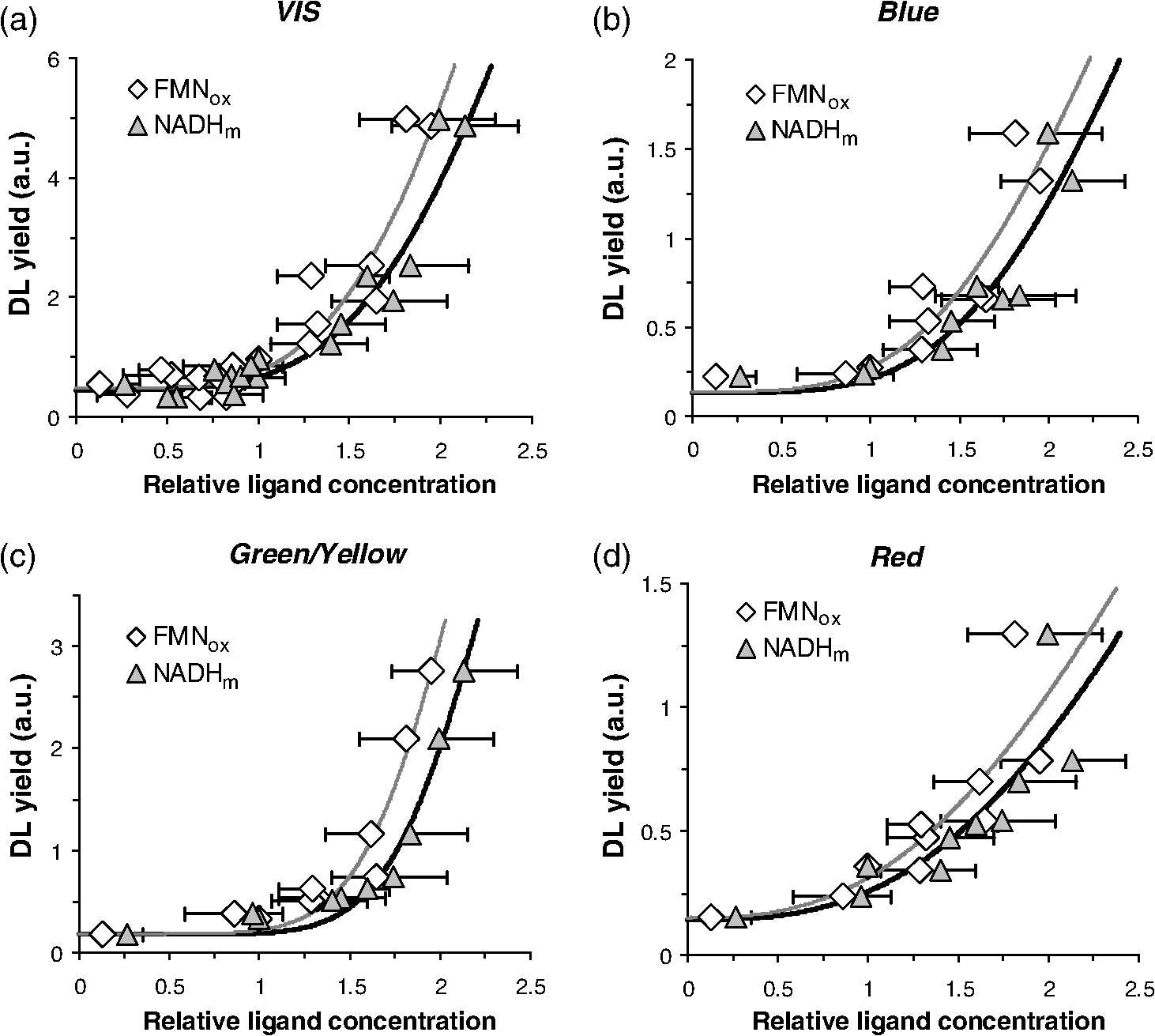 The total yield of delayed emission of blue, green/yellow, or red light exhibited a qualitatively similar dependence on and [] [Figs. 9(b) to 9(d)]. However, the corresponding individual Hill coefficients varied from 3.7 to 4.0 (blue) to 6.9 to 7.4 (green/yellow) and 2.7 to 2.8 (red) (Table 2). As also discussed below, all these figures substantiate the idea that the dominant structural arrangement of complex I in Jurkat cells is the dimeric form, which binds cooperatively to two ROT molecules, and that a significant contribution also comes from tetrameric complex I, in which at least three (most likely four) molecules of ROT bind to ROT site 1 in a cooperative manner. Table 2Fit-derived parameters of the DL-yield dependence on relative [NADH]m and [FMNox]. The DL yield in the indicated spectral domains was calculated in the time interval of 11 μs to 10 ms.
From a quick inspection of the spectral DL domains, we can gain some relevant insights concerning the relative frequency of complex I dimers and tetramers. Thus, the DL data indicate that green/yellow light emission (presumably associated with tetrameric complex I) contributes 39%, and blue- and red-light emission (associated with dimeric complex I) contributes 61% to total DL. So, our results suggest that and 40% of complex I oligomers in Jurkat cells are in dimeric and tetrameric form, respectively. In conclusion, based on the main current findings that (1) DL is closely related to the level of , which is found primarily in the mitochondria,65,90 (2) DL is also linked to the level of NADH, the substrate of mitochondrial complex I, (3) ROT, a specific inhibitor of complex I, affected DL considerably, and (4) MD and QC, which interact robustly with complex I, also affected DL significantly, all our results reinforce the idea62 that mitochondrial complex I plays a major role in the ultraweak photon-induced delayed photoemission in Jurkat cells. 4.DiscussionPerhaps the most physiologically relevant outcomes of our investigations derive from the finding that the DL yield depends in a Hill-like fashion on the level of NADH as well as that of , with apparent Hill coefficients of 4.3. In addition, spectral DL analysis indicated different degrees of cooperativity of both NADH and , reflected in differing Hill coefficients associated with blue-, green/yellow-, and red-light emission, namely 3.9, 7.2, and 2.8, respectively. In addition, fluorimetric measurements of NADH and levels indicated that ROT binding exhibits an apparent Hill coefficient of 2.0 to 2.4, suggesting that complex I functions mainly as a dimer in intact Jurkat cells. Based on these findings, we propose that blue and red DL is produced predominantly by dimeric complex I, whereas green/yellow DL arises mainly from tetrameric complex I. At first sight, these results could suggest a cooperative action of the NADH and NADPH sites of a complex I monomer,8 as well as cooperativity between both oxidized flavins [i.e., FMN-a and FMN-b (Ref. 8)], to generate DL-specific states, corroborated by the cooperative interaction between all monomers that are coupled within a complex I oligomeric form. In addition, a necessary condition would be that the concentrations of NADH and NADPH in the matrix are similar. Nevertheless, such cooperativity between sites situated at the two extremities of the hydrophilic sector of complex I has to involve a long-range interaction (which nevertheless has been documented91–93) and might reflect that in the particular complex I conformation induced by specific modulators like ROT, MD, and QC, the affinity of NADPH for its site is increased in the NADH-bound complex I. However, having in view that the concentration of NADPH in the matrix is generally much lower than that of NADH (reviewed in Ref. 64), a more plausible interpretation can be drawn from the idea of a tight interplay between complex I and nicotinamide nucleotide transhydrogenase (Nnt), a mitochondrial enzyme catalyzing the reaction . It is suggested8 that Nnt (a homodimer) can form a heteromer with complex I and thus regulate the ratio in the vicinity of the NADPH site of complex I (Fig. 1). By this mechanism, for example, Nnt cooperates with complex I in the attenuation of generation by complex I.8 It is thus conceivable that in situ, NADH binding to its site in complex I leads somehow to the stabilization of the complex I-Nnt heteromer, hence increasing the apparent rate (and affinity) of NADPH binding to its distal site on complex I. Thus, we propose that, in order for ROT-, MD-, or QC-bound complex I to generate DL, one NADH molecule must be bound to its site on complex I, one NADH molecule must associate with the Nnt monomer that is docked on the comer of complex I, and complex I and Nnt must form a tightly bound heteromer. Finally, in the same particular conformation of complex I, binding of NADH and similarly, NADPH to their corresponding sites enhance the rate of FMN-b and FMN-a oxidation, respectively. Therefore, an apparent Hill coefficient of 2 obtained for NADH per complex I monomer will be closely linked to an apparent Hill coefficient of 2 characteristic for . In the Nnt-complex I heterotetramer, the NADH binding sites situated on the two monomers of complex I, as well as the two NADH sites on the two Nnt monomers, must interact cooperatively. Thus, our results are consistent with the notion that in Jurkat cells, the main structural organization of mitochondrial complex I is most likely a supercomplex composed of one complex I dimer and one Nnt dimer. In favor of this, the concentrations of complex I and Nnt in bovine-heart submitochondrial particles have been found to be closely similar.8 At this point, it should be recalled that multiple dinucleotide [NAD(H) and NADP(H)] binding sites were identified within the sequence of complex I,94 and, consistent with this, five subunits isolated from complex I were found to bind NADH and/or NADPH.94 Steady-state kinetic studies of the transhydrogenase activity of complex I also indicated that a single dinucleotide-binding site is incompatible with the data.95 Moreover, cooperative binding of NADH in complex I, with an associated Hill coefficient of 2.3 (Ref. 18) or 2.6 (Ref. 96), has been reported by different groups. In addition, in complex I preparations where the Hill coefficient for NADH binding was 2.3, the corresponding Hill coefficient for ROT binding was found to be 1.1.18 We remark that the ratio of number of NADH molecules/number of ROT molecules bound to complex I derived in our study () is in good accord with these figures, but, based on the number of NADH and FMN molecules involved (two of each per bound molecule of ROT), suggests that the second active dinucleotide site must interact with the second flavin (i.e., FMN-a). However, at the moment, the only well-defined NADH-binding site of complex I remains that situated in the FMN-b containing 51 kDa subunit of the complex.92–94 Therefore, in the absence of conclusive knowledge of a functional NADH site that would be able to interact with FMN-a, we believe that the hypothesis of heteromeric Nnt-complex I coupling can reasonably explain our DL data. Moreover, the presumed ability of complex I to form oligomers in the native inner membrane of intact cell mitochondria is in line with recent evidences of the existence of large functional respirasome supercomplexes in various combinations of monomers, dimmers, or trimers of individual respiratory complexes with different stoichiometries.97,98 So, it is conceivable that in situ, complex I can form dimers, as also suggested by a number of previous studies,97–101 or even tetramers.102 To date, dimeric complex I has been detected in fungal mitochondria,98,99 and it has been proposed that such dimers may occur in organisms possessing alternative respiratory enzymes.98,99 It is thus possible that under the ROT blockage, the alternative oxidase is activated in Jurkat cells, hence increasing the likelihood of complex I aggregation. In favor of our hypothesis on the formation of complex I dimers or higher oligomers, we found in a different set of experiments that QC induces in Jurkat cells, long after drug removal, a persistent mitochondrial hyperpolarized state, which is inhibited by ROT in a biphasic manner, with corresponding Hill coefficients of 1.2 to 1.4 and 2.8 to 3.6, respectively (manuscript in preparation). Moreover, those measurements involved the fluorimetric detection of the fluorescent emission of a well-established fluorescent indicator, JC-1, not that of NADH or FMN. Hence, these latter results lend further support for the concept of cooperative interaction between two and three to four ROT binding sites, respectively. In addition, an independent nonfluorimetric assay conducted here on the basis of clonogenic survival determinations [Fig. 6(a)] also indicated cooperativity between at least two ROT binding sites, arguing against the idea that the current outcomes might reflect some potential fluorescence artifacts rather than the actual dependence of NADH and FMN levels on the ROT dose. In conclusion, the human leukemia Jurkat cell line appears to be an appealing candidate for further studies investigating the possible formation of complex I oligomers in mammalian cells. Our current results suggest that in Jurkat cells, complex I oligomers are formed, at least in the presence of ROT, QC, or MD, and may exist in either dimeric or tetrameric form, with an abundance ratio of . Approximately 50 to 90% of complex I is currently thought to be associated with dimeric complex III in supercomplexes that may also contain one to four subunits of complex IV.103,104 Interaction between complex I and complex III within the respirasomes was found to be essential to the activity and stability of complex I.97,101 Importantly, the existence of even larger supercomplexes containing multiple monomers of complexes I, III, and IV was unambiguously confirmed.104 It is therefore possible that our assay detected mixed responses originating from such supercomplexes assembled from oligomeric complex I and oligomeric complex III, having in view the peculiar effects of ROT and/or AA on flavin autofluorescence observed in our experiments. Knowledge of monomeric/oligomeric complex I specific distribution may be significant for the understanding of the molecular and functional properties of the respiratory complexes and supercomplexes in physiological or pathological conditions. Given the characteristic timescales of various electron transfer steps in complex I,16,17 we propose a minimal model consistent with the current DL data. UV photoexcitation of FMN generates excited FMN singlet states that may either decay to the ground state by prompt fluorescence105 or undergo intersystem crossing to long-lived triplet states,106 which can further relax to metastable intermediate states.106 In analogy with the Photosystem II case,58,60 the long lifetime of the triplet- or metastable-state species allows a series of photochemical reactions to occur in complex I and produces secondary excitations, thus giving rise to the ultraweak photon-induced delayed photoemission. The currently described states emitting blue or green/yellow light are most likely excited states of NADH or , respectively, that are produced after a series of forward and backward electron transfer steps reaching up to the Fe/S center N4 [Fig. 10(A)]. A necessary condition for the two sequences shown in Figs. 10(Aa) and 10(Ab) is that the primary oxidized molecule does not dissociate from complex I during the entire time interval. Indeed, the average time for which remains attached to complex I (, Refs. 16 and 17) is much larger than the overall lifetime of 29.3 μs estimated for the domain of the blue light emission traces (not shown). Next, the states emitting blue (or green/yellow) light may represent excited states of NADH (or ) regenerated via backward charge recombination after the primary sequence of forward transfer steps reaching the center N2 [Fig. 10(Ba)]. The appearance of states probably involves charge transfer from center N1b to N5 and backward, as well as from center N1a to N3 [Fig. 10(C)]. In view of the similarities with the other spectral components (e.g., Fig. 8), delayed emission of red light appears as well to be closely linked to complex I. In consequence, we propose that red DL is most likely produced via dimol photoemission generated by two colliding molecules of singlet oxygen (). Dimol luminescence can be observed at the characteristic wavelengths of 579, 634, and 703 nm,107,108 with main emission at 634 nm. Thus, increased levels of superoxide produced by the semiflavone at the FMN-b [Figs. 10(Ac), 10(Ad), 10(Bb), and 10(Ce)] or FMN-a site [Figs. 10(Bd) and 10(Bh)], or by the ubisemiquinone at the Q site [Figs. 10(Bc), 10(Cd), 10(Cf), 10(Cg), and 10(Ci)] could lead, particularly in the presence of superoxide dismutase, to the generation of significant levels of singlet oxygen109 and consequent dimol photoemission. We should note that our results differ from those of Mik et al.51,52 who found that ROT and other mitochondrial respiration inhibitors decrease the delayed (red) luminescence of endogenous protoporphyrin IX (PpIX), which is synthesized inside the mitochondria. ROT inhibition of PpIX red DL appeared to be a process mediated by increased levels of mitochondrial .51 However, the experimental design used therein was based on the application of a PpIX precursor [5-aminolevulinic acid hydrochloride (ALA)] to enhance the intracellular PpIX concentration. In the absence of ALA, red DL of HeLa cells was virtually undetectable [Fig. 4(a) in Ref. 51]. Under our conditions (excitation at 337 nm, versus 405 nm in Ref. 51 or 510 nm in Ref. 52), we observed a strong enhancement of red DL by ROT, which could suggest that, at least in Jurkat cells, singlet oxygen is the likely dominant generator of red DL observed upon excitation at 337 nm. On this basis, our current DL data suggest that the average time elapsed since the generation of semiflavone at FMN-b until the collision of the emerging with a second singlet oxygen situated in close vicinity is 13.3 μs [Fig. 10(Ac)]. For simplicity, we also assume that the other two sites of superoxide generation in complex I, namely FMN-a and Q, exhibit the same kinetics of dimol photoemission, with an overall time of electron transfer from semiflavone/semiquinone to molecular oxygen, dimol production and radiative decay, of 13.3 μs. Moreover, our DL data are best explained by assuming that center N2 is reduced by NADH within 90 μs, in accord with previous reports,16 which provides a characteristic time of 54 μs required for the electron transfer from center N6b to N2 [Fig. 10(Ba)]. It is worth noting that the two values derived above explain very well the dominant kinetic component of both blue and green/yellow (140 μs derived from DL emission curves, not shown), as well as red light emission from states (148 μs) produced via NADH reduction of N2, followed by reverse electron transfer to FMN-b and NADH [Figs. 10(Ba) and 10(Bb)]. However, we propose that the prominent component (129 μs) of red DL is produced via reduction of ubiquinone by the first electron of NADH received from center N2 (estimated time, 20 μs), generation of superoxide and then singlet oxygen dimol by the usual pathway [Fig. 10(Bc)]. In the presence of ROT, the ubisemiquinone becomes unstable, dissociates from the -site, and reduces to superoxide.20 In the absence of ROT, ubisemiquinone remains attached to the -site (for which it has a high affinity) and is further reduced to ubiquinol () by the second incoming electron from N2. , which cannot react with molecular oxygen because the reaction is spin forbidden,8 participates in electron transfer within the membrane domain and in proton translocation. In consequence, ROT will increase the red-light emission from the semiquinone radical produced at the Q-site via reduction by NADH. The slower component (390 μs) of red DL may be produced in a similar manner, via reduction of flavin FMN-a by NADH through center N2, and subsequent interaction with [Fig. 10(Bd)]. We estimated that in the presence of ROT, the mean time for electron transfer from reduced center N2 to FMN-a is 284 μs in situ, which suggests that the likelihood for N2 to reduce ubiquinone is 14 times higher than to reduce FMN-a. On the millisecond timescale, delayed red-light emission may involve electron transfer between centers N1b and N5, or transfer from center N1a to N3, and subsequent superoxide production at the FMN-b, FMN-a, or Q sites [Figs. 10(Cd), 10(Ce), 10(Cg), 10(Ch), and 10(Ci)]. In addition, we propose that the distinct red-DL component with decay time of 3.87 ms is associated with the reduction of ubiquinone by center N6a. Nevertheless, the long time required for this charge transfer reaction is compatible with a reduced likelihood of a process in which the long, flexible tail of ubiquinone manages to arrange itself inside the putative hydrophobic tunnel formed between the 49-kDa and PSST subunits of complex I (Ref. 8) (Fig. 1). Although the crystal structure of complex I isolated from two prokaryotic systems (E. coli, T. thermophilus) does not present clear evidence for this scenario,1,3,93 it is possible that it does not accurately reflect the constitution of mammalian complex I,110 which could present distinct particularities, especially in its native mitochondrial environment. Accordingly, our data suggest that both situations are possible; however the protrusion of ubiquinone into the hydrophobic tunnel up to cluster N6a appears to be a rather rare event, which could essentially be facilitated by the binding of ROT at site 1. Fluorimetric measurements performed in this study also demonstrated, contrary to expectations,65,81 a strong linear correlation between the mitochondrial level of NADH and that of in Jurkat cells treated with complex I targeting agents like ROT, MD, and QC. This rather unusual feature was observed previously in muscle tissue71 and human monocytes,72 where ROT was reported to increase flavoprotein emission. The correlation established here could suggest that in Jurkat cells, the ROT-, MD-, or QC-bound complex I accelerates the electron transfer from reduced FMN, either toward the neighbor Fe/S centers or backward, to the molecule, if the latter has not dissociated yet from FMN. In any case, assuming that the total level of mitochondrial flavins does not change significantly during the treatment, the observed increase in the fluorescence of at increasing levels of NADH definitely reflects an overall increase in the population (and relative dwell time) of the oxidized state of FMN and a corresponding decrease in the population (and relative dwell time) of the semi- or fully reduced states of FMN. Having in view that an NADH increase does not induce by itself an increase in the level of , as demonstrated by experiments employing the complex III inhibitor, AA [Figs. 7(c) and 7(g)], we infer that ROT, MD, and QC most likely induce a conformational change in complex I, in which the density of the population is directly related to the level of NADH in the matrix, and hence with the apparent residence time of at its binding site in complex I [since binding of NADH to the enzyme results in a long-range conformational change at the membrane interface (quinone-binding pocket),91–93 it is conceivable that the reverse situation is also possible]. A likely explanation may be that the binding of ROT or MD/QC to complex I in situ inhibits or stimulates, respectively, the dissociation of from complex I following reduction of FMN by NADH, and that associated favors the oxidized state of FMN in this particular conformation. However, since ROT does not affect the reduction kinetics of neither of the Fe/S clusters,8,100 it seems more likely that the semiflavin radical , which is produced rapidly after reduction of center N3a by , dislocates within the FMN binding cavity and produces a conformational change of the protein,87 which would allow access to molecular oxygen (even in the presence of in the substrate binding cleft), leading to subsequent production of superoxide. Regenerated can then rebind with high affinity to its site. Moreover, high levels of NADH appear to stimulate the dissociation of from the FMN site,87 which lends further support for our interpretation. Thus, our data suggest that ROT increases, whereas MD and QC decrease, the probability of reduction by via modulation of the NADH level and also by conformational facilitation of the NADH effect on . In addition, since both QC and MD decrease and [] and also have closely similar effects on DL (Ref. 62), it is expected that both agents share a common binding site and operate by similar mechanisms at the level of complex I in Jurkat cells. Reports that QC can act as a coenzyme Q-mimetic molecule38 or decrease the ROS production by complex I (Ref. 39) further substantiate the current proposal that MD and QC decrease the affinity of for the reduced form of complex I (the apparent under normal conditions87) by stimulating the dissociation of from reduced complex I. In turn, the vacancy in the NADH site hampers the detachment of , so the inability of the flavin radical to access dioxygen leads to decreased superoxide production. The observation that ROT exerts opposed effects in production by mitochondria, although its site is situated in the same hydrophobic cavity at the matrix/membrane interface,39 is also in agreement with our results. QC and MD are likely to inhibit the accessibility of ubiquinone to both Fe/S centers N2 and N6a, as well as the reduction of N2 by FMN-a, hence explaining the considerable decrease of DL observed especially on the and timescales.62 Interestingly, in the rat brain and heart mitochondrial preparations of Lagoa and collaborators, QC exercised its inhibitory effects on complex I without affecting the respiration (oxygen consumption) rate,39 suggesting that QC is able to both receive electrons from N2 and redirect them to without the destabilization of the intermediary radical, thereby acting as a substitute for ubiquinone.39 However, since MD produces superoxide at complex I by forming a semiquinone,27,35 presumably after accepting one electron from center N2, we infer that the main mechanism responsible for the observed effects of MD and QC on DL are caused by the inhibition of (1) semiflavin lifetime (indirectly via an mediated effect), (2) ubiquinone reduction at center N6a, and (3) N2 reduction by FMN-a, whereas ROT enhances DL by stimulating all these processes and also the backward electron transfer from center N2 to FMN-b (due to an increased pool of electrons dwelling inside complex I). In addition, having in view that the rate-limiting step that defines the enzymatic activity of complex I is the dissociation of from its site, which has a typical lifetime of under normal conditions,16,17 our results support the idea that ROT inhibits, whereas MD and QC stimulate, complex I activity by inhibiting or stimulating the dissociation of . 5.ConclusionWe present evidence that the ultraweak photon-induced delayed photoemission of intact Jurkat cells originates mainly from mitochondrial complex I, which appears to function predominantly as a dimer (with a relative frequency of ) and less frequently as a tetramer (with a relative frequency of ) in this cell type. Complex I oligomers appear to exhibit cooperative interaction between monomers at the level of the ROT site 1, NADH/NADPH sites, and FMN-b/FMN-a sites. Moreover, in individual monomers, both pairs of pyridine nucleotide (NADH/NADPH) binding sites and flavin (FMN-b/FMN-a) binding sites, which are situated at the two extremities of the extramembranous domain of complex I, display strong cooperativity in the binding of their specific ligands. In addition, the timescales of various electron transfer steps involving the formation of flavin and ubisemiquinone radicals with subsequent production of superoxide can be estimated from delayed red-light emission. All these findings raise the attractive possibility that DL spectroscopy could be used as a reliable, sensitive, and robust technique to probe electron flow within complex I and gain valuable insights into the structural and functional organization of this respiratory complex in situ. Future developments toward clinical diagnosis of mitochondrial disorders or cancer can be envisioned. AcknowledgmentsThis work was supported by a grant of the Romanian National Authority for Scientific Research, CNCS-UEFISCDI, project number PN-II-ID-PCE-2011-3-0800. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. ReferencesR. G. EfremovR. BaradaranL. A. Sazanov,
“The architecture of respiratory complex I,”
Nature, 465
(7297), 441
–445
(2010). http://dx.doi.org/10.1038/nature09066 NATUAS 0028-0836 Google Scholar
T. Ohnishiet al.,
“EPR detection of two protein-associated ubiquinone components (SQNf and SQNs) in the membrane in situ and in proteoliposomes of isolated bovine heart complex I,”
Biochim. Biophys. Acta, 1817
(10), 1803
–1809
(2012). http://dx.doi.org/10.1016/j.bbabio.2012.03.032 BBACAQ 0006-3002 Google Scholar
R. Baradaranet al.,
“Crystal structure of the entire respiratory complex I,”
Nature, 494
(7438), 443
–448
(2013). http://dx.doi.org/10.1038/nature11871 NATUAS 0028-0836 Google Scholar
T. OhnishiS. T. Ohnishi,
“A new trend in the complex I research field,”
Biol. Chem., 394
(5), 677
–683
(2013). http://dx.doi.org/10.1515/hsz-2013-0117 BICHF3 1431-6730 Google Scholar
S. P. J. AlbrachtR. Hedderich,
“Learning from hydrogenases: location of a proton pump and of a second FMN in bovine NADH-ubiquinone oxidoreductase (complex I),”
FEBS Lett., 485
(1), 1
–6
(2000). http://dx.doi.org/10.1016/S0014-5793(00)02172-4 FEBLAL 0014-5793 Google Scholar
S. P. J. Albracht,
“The reaction of NADPH with bovine mitochondrial NADH:ubiquinone oxidoreductase revisited, I. Proposed consequences for electron transfer in the enzyme,”
J. Bioenerg. Biomembr., 42
(4), 261
–278
(2010). http://dx.doi.org/10.1007/s10863-010-9301-z JBBID4 0145-479X Google Scholar
S. P. J. Albracht,
“The reaction of NADPH with bovine mitochondrial NADH:ubiquinone oxidoreductase revisited, II. Comparison of the proposed working hypothesis with literature data,”
J. Bioenerg. Biomembr., 42
(4), 279
–292
(2010). http://dx.doi.org/10.1007/s10863-010-9302-y JBBID4 0145-479X Google Scholar
S. P. J. AlbrachtA. J. MeijerJ. Rydström,
“Mammalian NADH:ubiquinone oxidoreductase (complex I) and nicotinamide nucleotide transhydrogenase (Nnt) together regulate the mitochondrial production of —implications for their role in disease, especially cancer,”
J. Bioenerg. Biomembr., 43
(5), 541
–564
(2011). http://dx.doi.org/10.1007/s10863-011-9381-4 JBBID4 0145-479X Google Scholar
S. Fleischeret al.,
“Lipid composition of mitochondria from bovine heart, liver, and kidney,”
J. Lipid Res., 8
(3), 170
–180
(1967). JLPRAW 0022-2275 Google Scholar
M. TurunenJ. OlssonG. Dallner,
“Metabolism and function of coenzyme Q,”
Biochim. Biophys. Acta, 1660
(1–2), 171
–199
(2004). http://dx.doi.org/10.1016/j.bbamem.2003.11.012 BBACAQ 0006-3002 Google Scholar
Y. Nakashimaet al.,
“The second coenzyme Q1 binding site of bovine heart NADH: coenzyme Q oxidoreductase,”
J. Bioenerg. Biomembr., 34
(2), 89
–94
(2002). http://dx.doi.org/10.1023/A:1015119808009 JBBID4 0145-479X Google Scholar
M. Muraiet al.,
“Characterization of the inhibitor binding site in mitochondrial NADH-ubiquinone oxidoreductase by photoaffinity labeling using a quinazoline-type inhibitor,”
Biochemistry, 48
(4), 688
–698
(2009). http://dx.doi.org/10.1021/bi8019977 BIORAK 0006-2979 Google Scholar
M. Muraiet al.,
“Exploring interactions between the 49 kDa and ND1 subunits in mitochondrial NADH-ubiquinone oxidoreductase (complex I) by photoaffinity labeling,”
Biochemistry, 50
(32), 6901
–6908
(2011). http://dx.doi.org/10.1021/bi200883c BIORAK 0006-2979 Google Scholar
T. Clasonet al.,
“The structure of eukaryotic and prokaryotic complex I,”
J. Struct. Biol., 169
(1), 81
–88
(2010). http://dx.doi.org/10.1016/j.jsb.2009.08.017 JSBIEM 1047-8477 Google Scholar
T. OhnishiE. Nakamaru-OgisoS. T. Ohnishi,
“A new hypothesis on the simultaneous direct and indirect proton pump mechanisms in NADH-quinone oxidoreductase (complex I),”
FEBS Lett., 584
(19), 4131
–4137
(2010). http://dx.doi.org/10.1016/j.febslet.2010.08.039 FEBLAL 0014-5793 Google Scholar
M. L. Verkhovskayaet al.,
“Real-time electron transfer in respiratory complex I,”
Proc. Natl. Acad. Sci. U. S. A., 105
(10), 3763
–3767
(2008). http://dx.doi.org/10.1073/pnas.0711249105 PNASA6 0027-8424 Google Scholar
S. RansacC. ArnarezJ. P. Mazat,
“The flitting of electrons in complex I: a stochastic approach,”
Biochim. Biophys. Acta, 1797
(6–7), 641
–648
(2010). http://dx.doi.org/10.1016/j.bbabio.2010.03.011 BBACAQ 0006-3002 Google Scholar
H. SuzukiT. E. King,
“Evidence of an ubisemiquinone radical(s) from the NADH-ubiquinone reductase of the mitochondrial respiratory chain,”
J. Biol. Chem., 258
(1), 352
–358
(1983). JBCHA3 0021-9258 Google Scholar
S. Magnitskyet al.,
“EPR characterization of ubisemiquinones and iron-sulfur cluster N2, central components of the energy coupling in the NADH-ubiquinone oxidoreductase (complex I) in situ,”
J. Bioenerg. Biomembr., 34
(3), 193
–208
(2002). http://dx.doi.org/10.1023/A:1016083419979 JBBID4 0145-479X Google Scholar
S. T. Ohnishiet al.,
“A possible site of superoxide generation in the complex I segment of rat heart mitochondria,”
J. Bioenerg. Biomembr., 37
(1), 1
–15
(2005). http://dx.doi.org/10.1007/s10863-005-4117-y JBBID4 0145-479X Google Scholar
S. Dröseet al.,
“Functional dissection of the proton pumping modules of mitochondrial complex I,”
PLoS Biol., 9
(8), e1001128
(2011). http://dx.doi.org/10.1371/journal.pbio.1001128 1544-9173 Google Scholar
S. Steimleet al.,
“Role of subunit NuoL for proton translocation by respiratory complex I,”
Biochemistry, 50
(16), 3386
–3393
(2011). http://dx.doi.org/10.1021/bi200264q BIORAK 0006-2979 Google Scholar
M. Narayananet al.,
“Semiquinone and cluster N6 signals in His-tagged proton translocating NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli,”
J. Biol. Chem., 288
(20), 14310
–14319
(2013). http://dx.doi.org/10.1074/jbc.M113.467803 JBCHA3 0021-9258 Google Scholar
D. E. KoeppeR. J. Miller,
“Oxidation of reduced nicotinamide adenine dinucleotide phosphate by isolated corn mitochondria,”
Plant Physiol., 49
(3), 353
–357
(1972). http://dx.doi.org/10.1104/pp.49.3.353 PLPHAY 0032-0889 Google Scholar
Y. HatefiW. G. Hanstein,
“Interactions of reduced and oxidized triphosphopyridine nucleotides with the electron-transport system of bovine heart mitochondria,”
Biochemistry, 12
(18), 3515
–3522
(1973). http://dx.doi.org/10.1021/bi00742a026 BIORAK 0006-2979 Google Scholar
Y. M. GalanteY. Hatefi,
“Purification and molecular and enzymatic properties of mitochondrial NADH dehydrogenase,”
Arch. Biochem. Biophys., 192
(2), 559
–568
(1979). http://dx.doi.org/10.1016/0003-9861(79)90126-7 ABBIA4 0003-9861 Google Scholar
X. XuE. A. Arriaga,
“Qualitative determination of superoxide release at both sides of the mitochondrial inner membrane by capillary electrophoretic analysis of the oxidation products of triphenylphosphonium hydroethidine,”
Free Rad. Biol. Med., 46
(7), 905
–913
(2009). http://dx.doi.org/10.1016/j.freeradbiomed.2008.12.019 FRBMEH 0891-5849 Google Scholar
N. Liet al.,
“Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production,”
J. Biol. Chem., 278
(10), 8516
–8525
(2003). http://dx.doi.org/10.1074/jbc.M210432200 JBCHA3 0021-9258 Google Scholar
V. G. Grivennikovaet al.,
“Interaction of the mitochondrial NADH-ubiquinone reductase with rotenone as related to the enzyme active/inactive transition,”
Biochim. Biophys. Acta, 1319
(2–3), 223
–232
(1997). http://dx.doi.org/10.1016/S0005-2728(96)00163-6 BBACAQ 0006-3002 Google Scholar
J. S. IsenbergJ. E. Klaunig,
“Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells,”
Toxicol. Sci., 53
(2), 340
–351
(2000). http://dx.doi.org/10.1093/toxsci/53.2.340 TOSCF2 1096-0929 Google Scholar
W. Yinet al.,
“Plasma membrane depolarization and Na,K-ATPase impairment induced by mitochondrial toxins augment leukemia cell apoptosis via a novel mitochondrial amplification mechanism,”
Biochem. Pharmacol., 78
(2), 191
–202
(2009). http://dx.doi.org/10.1016/j.bcp.2009.03.025 BCPCA6 0006-2952 Google Scholar
S. Matznoet al.,
“An attempt to evaluate the effect of vitamin K3 using as an enhancer of anticancer agents,”
Biol. Pharm. Bull., 31
(6), 1270
–1273
(2008). http://dx.doi.org/10.1248/bpb.31.1270 BPBLEO 0918-6158 Google Scholar
I. LauxA. Nel,
“Evidence that oxidative stress-induced apoptosis by menadione involves Fas-dependent and Fas-independent pathways,”
Clin. Immunol., 101
(3), 335
–344
(2001). http://dx.doi.org/10.1006/clim.2001.5129 1521-6616 Google Scholar
J. J. Brièreet al.,
“Quinone analogues regulate mitochondrial substrate competitive oxidation,”
Biochem. Biophys. Res. Commun., 316
(4), 1138
–1142
(2004). http://dx.doi.org/10.1016/j.bbrc.2004.03.002 BBRCA9 0006-291X Google Scholar
M. FloreaniF. Carpenedo,
“One- and two-electron reduction of menadione in guinea-pig and rat cardiac tissue,”
Gen. Pharmacol., 23
(4), 757
–762
(1992). http://dx.doi.org/10.1016/0306-3623(92)90162-D GEPHDP 0306-3623 Google Scholar
P. J. O’Brien,
“Molecular mechanisms of quinone cytotoxicity,”
Chem. Biol. Interact., 80
(1), 1
–41
(1991). http://dx.doi.org/10.1016/0009-2797(91)90029-7 CBINA8 0009-2797 Google Scholar
J. Shenet al.,
“Oxygen consumption rates and oxygen concentration in Molt-4 cells and their mtDNA depleted () mutants,”
Biophys. J., 84
(2), 1291
–1298
(2003). http://dx.doi.org/10.1016/S0006-3495(03)74944-3 BIOJAU 0006-3495 Google Scholar
C. Sandoval-Acuñaet al.,
“Inhibition of mitochondrial complex I by various non-steroidal anti-inflammatory drugs and its protection by quercetin via a coenzyme Q-like action,”
Chem. Biol. Interact., 199
(1), 18
–28
(2012). http://dx.doi.org/10.1016/j.cbi.2012.05.006 CBINA8 0009-2797 Google Scholar
R. Lagoaet al.,
“Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria,”
Biochim. Biophys. Acta, 1807
(12), 1562
–1572
(2011). http://dx.doi.org/10.1016/j.bbabio.2011.09.022 BBACAQ 0006-3002 Google Scholar
C. Carrasco-Pozoet al.,
“Differential protective effects of quercetin, resveratrol, rutin and epigallocatechin gallate against mitochondrial dysfunction induced by indomethacin in Caco-2 cells,”
Chem. Biol. Interact., 195
(3), 199
–205
(2012). http://dx.doi.org/10.1016/j.cbi.2011.12.007 CBINA8 0009-2797 Google Scholar
I. Baranet al.,
“Fluorescence properties of quercetin in human leukemia Jurkat T-cells,”
Rom. J. Phys., 56
(3–4), 388
–398
(2011). RJPHEC 1221-146X Google Scholar
I. Baranet al.,
“Detailed analysis of apoptosis and delayed luminescence of human leukemia Jurkat T cells after proton-irradiation and treatments with oxidant agents and flavonoids,”
Oxid. Med. Cell. Longev., 2012 498914
(2012). http://dx.doi.org/10.1155/2012/498914 OMCLAB 1942-0994 Google Scholar
I. BaranE. KatonaC. Ganea,
“Quercetin as a fluorescent probe for the ryanodine receptor activity in Jurkat cells,”
Pflug. Arch. Eur. J. Phy., 465
(8), 1101
–1119
(2013). http://dx.doi.org/10.1007/s00424-013-1235-y PFLABK 0031-6768 Google Scholar
D. J. Dortaet al.,
“The interaction of flavonoids with mitochondria: effects on energetic processes,”
Chem. Biol. Interact., 152
(2–3), 67
–78
(2005). http://dx.doi.org/10.1016/j.cbi.2005.02.004 CBINA8 0009-2797 Google Scholar
M. Fioraniet al.,
“Mitochondria accumulate large amounts of quercetin: prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid,”
J. Nutr. Biochem., 21
(5), 397
–404
(2010). http://dx.doi.org/10.1016/j.jnutbio.2009.01.014 JNBIEL 0955-2863 Google Scholar
U. De Marchiet al.,
“Quercetin can act either as an inhibitor or an inducer of the mitochondrial permeability transition pore: a demonstration of the ambivalent redox character of polyphenols,”
Biochim. Biophys. Acta, 1787
(12), 1425
–1432
(2009). http://dx.doi.org/10.1016/j.bbabio.2009.06.002 BBACAQ 0006-3002 Google Scholar
M. A. Ortneret al.,
“Time gated fluorescence spectroscopy in Barrett’s oesophagus,”
Gut, 52
(1), 28
–33
(2003). http://dx.doi.org/10.1136/gut.52.1.28 GUTTAK 0017-5749 Google Scholar
F. Musumeciet al.,
“Spectral analysis of laser-induced ultraweak delayed luminescence in cultured normal and tumor human cells: temperature dependence,”
J. Photochem. Photobiol. B, 79
(2), 93
–99
(2005). http://dx.doi.org/10.1016/j.jphotobiol.2004.12.002 JPPBEG 1011-1344 Google Scholar
H. W. Kimet al.,
“Spontaneous photon emission and delayed luminescence of two types of human lung cancer tissues: adenocarcinoma and squamous cell carcinoma,”
Cancer Lett., 229
(2), 283
–289
(2005). http://dx.doi.org/10.1016/j.canlet.2005.04.038 CALEDQ 0304-3835 Google Scholar
W. Kemmneret al.,
“Silencing of human ferrochelatase causes abundant protoporphyrin-IX accumulation in colon cancer,”
FASEB J., 22
(2), 500
–509
(2008). http://dx.doi.org/10.1096/fj.07-8888com FAJOEC 0892-6638 Google Scholar
E. G. Miket al.,
“Mitochondrial measured by delayed fluorescence of endogenous protoporphyrin IX,”
Nat. Methods, 3
(11), 939
–945
(2006). http://dx.doi.org/10.1038/nmeth940 1548-7091 Google Scholar
E. G. Miket al.,
“In vivo mitochondrial oxygen tension measured by a delayed fluorescence lifetime technique,”
Biophys. J., 95
(8), 3977
–3990
(2008). http://dx.doi.org/10.1529/biophysj.107.126094 BIOJAU 0006-3495 Google Scholar
P. Felkeret al.,
“Effects of electron transport inhibitors on millisecond delayed light emission from chloroplasts,”
Biochim. Biophys. Acta, 325
(1), 193
–196
(1973). http://dx.doi.org/10.1016/0005-2728(73)90166-7 BBACAQ 0006-3002 Google Scholar
F. A. Poppet al.,
“Biophoton emission. New evidence for coherence and DNA as source,”
Cell Biophys., 6
(1), 33
–52
(1984). CBIODE 0163-4992 Google Scholar
J. Slawinski,
“Luminescence research and its relation to ultraweak cell radiation,”
Experientia, 44
(7), 559
–571
(1988). http://dx.doi.org/10.1007/BF01953303 EXPEAM 0014-4754 Google Scholar
E. HidegM. KobayashiH. Inaba,
“Spontaneous ultraweak light emission from respiring spinach leaf mitochondria,”
Biochim. Biophys. Acta, 1098
(1), 27
–31
(1991). http://dx.doi.org/10.1016/0005-2728(91)90005-9 BBACAQ 0006-3002 Google Scholar
S. Tudiscoet al.,
“ARETUSA—advanced research equipment for fast ultraweak luminescence analysis: new developments,”
Nucl. Instrum. Methods Phys. Res. A, 518
(1–2), 463
–464
(2004). http://dx.doi.org/10.1016/j.nima.2003.11.056 NIMAER 0168-9002 Google Scholar
V. Goltsevet al.,
“Kinetics of delayed chlorophyll a fluorescence registered in milliseconds time range,”
Photosynth. Res., 84
(1–3), 209
–215
(2005). http://dx.doi.org/10.1007/s11120-004-6432-2 PHRSDI 0166-8595 Google Scholar
M. Katsumataet al.,
“New feature of delayed luminescence: preillumination-induced concavity and convexity in delayed luminescence decay curve in the green alga Pseudokirchneriella subcapitata,”
J. Photochem. Photobiol. B, 90
(3), 152
–162
(2008). http://dx.doi.org/10.1016/j.jphotobiol.2007.12.005 JPPBEG 1011-1344 Google Scholar
Y. GuoJ. Tan,
“A kinetic model structure for delayed fluorescence from plants,”
BioSystems, 95
(2), 98
–103
(2009). http://dx.doi.org/10.1016/j.biosystems.2008.08.002 BSYMBO 0303-2647 Google Scholar
I. Baranet al.,
“Effects of nocodazole and ionizing radiation on cell proliferation and delayed luminescence,”
Rom. J. Phys., 54
(5–6), 557
–567
(2009). RJPHEC 1221-146X Google Scholar
I. Baranet al.,
“Effects of menadione, hydrogen peroxide and quercetin on apoptosis and delayed luminescence of human leukemia Jurkat T-cells,”
Cell Biochem. Biophys., 58
(3), 169
–179
(2010). http://dx.doi.org/10.1007/s12013-010-9104-1 1085-9195 Google Scholar
A. Scordinoet al.,
“Influence of the presence of atrazine in water on the in-vivo delayed luminescence of Acetabularia acetabulum,”
J. Photochem. Photobiol. B: Biol., 32
(1–2), 11
–17
(1996). http://dx.doi.org/10.1016/1011-1344(95)07213-6 JPPBEG 1011-1344 Google Scholar
A. MayevskyG. G. Rogatsky,
“Mitochondrial function in vivo evaluated by NADH fluorescence: from animal models to human studies,”
Am. J. Physiol. Cell Physiol., 292
(2), C615
–C640
(2007). http://dx.doi.org/10.1152/ajpcell.00249.2006 0363-6143 Google Scholar
L. D. GaspersA. P. Thomas,
“Calcium-dependent activation of mitochondrial metabolism in mammalian cells,”
Methods, 46
(3), 224
–232
(2008). http://dx.doi.org/10.1016/j.ymeth.2008.09.012 MTHDE9 1046-2023 Google Scholar
R. C. Bensonet al.,
“Cellular autofluorescence—is it due to flavins?,”
J. Histochem. Cytochem., 27
(1), 44
–48
(1979). http://dx.doi.org/10.1177/27.1.438504 JHCYAS 0022-1554 Google Scholar
R. BrandesD. M. Bers,
“Simultaneous measurements of mitochondrial NADH and during increased work in intact rat heart trabeculae,”
Biophys. J., 83
(2), 587
–604
(2002). http://dx.doi.org/10.1016/S0006-3495(02)75194-1 BIOJAU 0006-3495 Google Scholar
M. L. Riesset al.,
“Reduced reactive species formation and preserved mitochondrial NADH and [] levels during short-term 17°C ischemia in intact hearts,”
Cardiovasc. Res., 61
(3), 580
–590
(2004). http://dx.doi.org/10.1016/j.cardiores.2003.09.016 CVREAU 0008-6363 Google Scholar
J. EngR. M. LynchR. S. Balaban,
“Nicotinamide adenine dinucleotide fluorescence spectroscopy and imaging of isolated cardiac myocytes,”
Biophys. J., 55
(4), 621
–630
(1989). http://dx.doi.org/10.1016/S0006-3495(89)82859-0 BIOJAU 0006-3495 Google Scholar
I. M. Møller,
“Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species,”
Ann. Rev. Plant Physiol. Plant Mol. Biol., 52 561
–591
(2001). http://dx.doi.org/10.1146/annurev.arplant.52.1.561 ARPBEX 1040-2519 Google Scholar
A. V. Kuznetsovet al.,
“Functional imaging of mitochondria in saponin-permeabilized mice muscle fibers,”
J. Cell Biol., 140
(5), 1091
–1099
(1998). http://dx.doi.org/10.1083/jcb.140.5.1091 JCLBA3 0021-9525 Google Scholar
A. KindzelskiiH. R. Petty,
“Fluorescence spectroscopic detection of mitochondrial flavoproteins redox oscillations and transient reduction of the NADPH oxidase-associated flavoprotein in leukocytes,”
Eur. Biophys. J., 33
(4), 291
–299
(2004). http://dx.doi.org/10.1007/s00249-003-0361-4 EBJOE8 0175-7571 Google Scholar
W. S. Kunz,
“Spectral properties of fluorescent flavoproteins of isolated rat liver mitochondria,”
FEBS Lett., 195
(1–2), 92
–96
(1986). http://dx.doi.org/10.1016/0014-5793(86)80137-5 FEBLAL 0014-5793 Google Scholar
D. Chorvat Jr.et al.,
“Spectral unmixing of flavin autofluorescence components in cardiac myocytes,”
Biophys. J., 89
(6), L55
–57
(2005). http://dx.doi.org/10.1529/biophysj.105.073866 BIOJAU 0006-3495 Google Scholar
J. V. RocheleauW. S. HeadD. W. Piston,
“Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response,”
J. Biol. Chem., 279
(30), 31780
–31787
(2004). http://dx.doi.org/10.1074/jbc.M314005200 JBCHA3 0021-9258 Google Scholar
A. Höhnet al.,
“Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells,”
Free Radic. Biol. Med., 48
(8), 1100
–1108
(2010). http://dx.doi.org/10.1016/j.freeradbiomed.2010.01.030 FRBMEH 0891-5849 Google Scholar
I. GeorgakoudiK. P. Quinn,
“Optical imaging using endogenous contrast to assess metabolic state,”
Annu. Rev. Biomed. Eng., 14 351
–367
(2012). http://dx.doi.org/10.1146/annurev-bioeng-071811-150108 ARBEF7 1523-9829 Google Scholar
A. J. W. G. Visseret al.,
“Time-resolved fluorescence studies of flavodoxin,”
FEBS Lett., 224
(2), 406
–410
(1987). http://dx.doi.org/10.1016/0014-5793(87)80493-3 FEBLAL 0014-5793 Google Scholar
J. A. GondalW. M. Anderson,
“The molecular morphology of bovine heart mitochondrial NADH-ubiquinone reductase. Cross-linking with dithiobis(succinimidyl propionate),”
J. Biol. Chem., 260
(10), 5931
–5935
(1985). JBCHA3 0021-9258 Google Scholar
A. J. Lambertet al.,
“Low complex I content explains the low hydrogen peroxide production rate of heart mitochondria from the long-lived pigeon, Columba livia,”
Aging Cell, 9
(1), 78
–91
(2010). http://dx.doi.org/10.1111/ace.2010.9.issue-1 ACGECQ 1474-9718 Google Scholar
C. W. Shuttleworth,
“Use of NAD(P)H and flavoprotein autofluorescence transients to probe neuron and astrocyte responses to synaptic activation,”
Neurochem. Int., 56
(3), 379
–386
(2010). http://dx.doi.org/10.1016/j.neuint.2009.12.015 NEUIDS 0197-0186 Google Scholar
G. A. Martenset al.,
“Glucose suppresses superoxide generation in metabolically responsive pancreatic cells,”
J. Biol. Chem., 280
(21), 20389
–20396
(2005). http://dx.doi.org/10.1074/jbc.M411869200 JBCHA3 0021-9258 Google Scholar
X. Yuet al.,
“Isradipine prevents rotenone-induced intracellular calcium rise that accelerates senescence in human neuroblastoma SH-SY5Y cells,”
Neurosci., 246 243
–253
(2013). http://dx.doi.org/10.1016/j.neuroscience.2013.04.062 NROSFJ 1073-8584 Google Scholar
M. MoslehiR. Yazdanparast,
“Protective effects of flavonoid baicalein against menadione-induced damage in SK-N-MC cells,”
CellBio, 2
(2), 35
–44
(2013). http://dx.doi.org/10.4236/cellbio.2013.22005 CELLF9 2325- 7792 Google Scholar
D. N. RomashkoE. MarbanB. O’Rourke,
“Subcellular metabolic transients and mitochondrial redox waves in heart cells,”
Proc. Natl. Acad. Sci. U.S.A., 95
(4), 1618
–1623
(1998). http://dx.doi.org/10.1073/pnas.95.4.1618 PNASA6 0027-8424 Google Scholar
K. J. BucklerR. D. Vaughan-Jones,
“Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells,”
J. Physiol., 513
(3), 819
–833
(1998). http://dx.doi.org/10.1111/tjp.1998.513.issue-3 JPHYA7 0022-3751 Google Scholar
A. D. Vinogradov,
“ interaction with NADH:ubiquinone oxidoreductase (complex I),”
Biochim. Biophys. Acta, 1777
(7–8), 729
–734
(2008). http://dx.doi.org/10.1016/j.bbabio.2008.04.014 BBACAQ 0006-3002 Google Scholar
J. R. Barrioet al.,
“Flavin 1, N6-ethenoadenine dinucleotide: dynamic and static quenching of fluorescence,”
Proc. Natl. Acad. Sci. U. S. A., 70
(3), 941
–943
(1973). http://dx.doi.org/10.1073/pnas.70.3.941 PNASA6 0027-8424 Google Scholar
A. D. VinogradovV. G. Grivennikova,
“The mitochondrial complex I: progress in understanding of catalytic properties,”
IUBMB Life, 52
(3–5), 129
–134
(2001). http://dx.doi.org/10.1080/15216540152845920 IULIF8 1521-6543 Google Scholar
A. A. Heikal,
“Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies,”
Biomark. Med., 4
(2), 241
–263
(2010). http://dx.doi.org/10.2217/bmm.10.1 1752-0363 Google Scholar
N. Hanoet al.,
“Effect of the side chain structure of coenzyme Q on the steady state kinetics of bovine heart NADH:coenzyme Q oxidoreductase,”
J. Bioenerg. Biomembr., 35
(3), 257
–265
(2003). http://dx.doi.org/10.1023/A:1024663715931 JBBID4 0145-479X Google Scholar
L. A. Sazanov,
“Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain,”
Biochemistry, 46
(9), 2275
–2288
(2007). http://dx.doi.org/10.1021/bi602508x BIORAK 0006-2979 Google Scholar
J. M. BerrisfordL. A. Sazanov,
“Structural basis for the mechanism of respiratory complex I,”
J. Biol. Chem., 284
(43), 29773
–29783
(2009). http://dx.doi.org/10.1074/jbc.M109.032144 JBCHA3 0021-9258 Google Scholar
M. Yamaguchiet al.,
“The multiple nicotinamide nucleotide-binding subunits of bovine heart mitochondrial NADH:ubiquinone oxidoreductase (complex I),”
Eur. J. Biochem., 267
(2), 329
–336
(2000). http://dx.doi.org/10.1046/j.1432-1327.2000.00999.x EJBCAI 0014-2956 Google Scholar
N. V. ZakharovaT. V. ZharovaA. D. Vinogradov,
“Kinetics of transhydrogenase reaction catalyzed by the mitochondrial NADH-ubiquinone oxidoreductase (complex I) imply more than one catalytic nucleotide-binding sites,”
FEBS Lett., 444
(2–3), 211
–216
(1999). http://dx.doi.org/10.1016/S0014-5793(99)00062-9 FEBLAL 0014-5793 Google Scholar
G. DooijewaardE. C. Slater,
“Steady-state kinetics of high molecular weight (type-I) NADH dehydrogenase,”
Biochim. Biophys. Acta, 440
(1), 1
–15
(1976). http://dx.doi.org/10.1016/0005-2728(76)90109-2 BBACAQ 0006-3002 Google Scholar
A. Strohet al.,
“Assembly of respiratory complexes I, III, and IV into NADH oxidase supercomplex stabilizes complex I in Paracoccus denitrificans,”
J. Biol. Chem., 279
(6), 5000
–5007
(2004). http://dx.doi.org/10.1074/jbc.M309505200 JBCHA3 0021-9258 Google Scholar
I. Marqueset al.,
“Supramolecular organization of the respiratory chain in Neurospora crassa mitochondria,”
Eukaryot. Cell, 6
(12), 2391
–2405
(2007). http://dx.doi.org/10.1128/EC.00149-07 ECUEA2 1535 - 9786 Google Scholar
J. BrinkE. J. BoekemaE. F. J. Van Bruggen,
“Electron microscopy and image analysis of the complexes I and V of the mitochondrial respiratory chain,”
Electron Microsc. Rev., 1
(1), 175
–199
(1988). http://dx.doi.org/10.1016/0892-0354(88)90001-9 EMIREL 0892-0354 Google Scholar
R. van Belzenet al.,
“New evidence for the dimeric nature of NADH:Q oxidoreductase in bovine-heart submitochondrial particles,”
Biochim. Biophys. Acta, 1017
(2), 152
–159
(1990). http://dx.doi.org/10.1016/0005-2728(90)90146-U BBACAQ 0006-3002 Google Scholar
F. Krauseet al.,
“Supramolecular organization of cytochrome c oxidase- and alternative oxidase-dependent respiratory chains in the filamentous fungus Podospora anserine,”
J. Biol. Chem., 279
(25), 26453
–26461
(2004). http://dx.doi.org/10.1074/jbc.M402756200 JBCHA3 0021-9258 Google Scholar
E. J. Boekemaet al.,
“Structure of NADH:Q oxidoreductase from bovine heart mitochondria studied by electron microscopy,”
Biochim. Biophys. Acta, 679
(1), 7
–11
(1982). http://dx.doi.org/10.1016/0005-2728(82)90248-1 BBACAQ 0006-3002 Google Scholar
D. Moreno-Lastreset al.,
“Mitochondrial complex I plays an essential role in human respirasome assembly,”
Cell Metab., 15
(3), 324
–335
(2012). http://dx.doi.org/10.1016/j.cmet.2012.01.015 CMEEB5 1550-4131 Google Scholar
F. Krauseet al.,
“Respirasome-like supercomplexes in green leaf mitochondria of spinach,”
J. Biol. Chem., 279
(46), 48369
–48375
(2004). http://dx.doi.org/10.1074/jbc.M406085200 JBCHA3 0021-9258 Google Scholar
K. A. Fosteret al.,
“Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration,”
Progr. Neurobiol., 79
(3), 136
–171
(2006). http://dx.doi.org/10.1016/j.pneurobio.2006.07.001 PGNBA5 0301-0082 Google Scholar
T. E. Swartzet al.,
“The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin,”
J. Biol. Chem., 276
(39), 36493
–36500
(2001). http://dx.doi.org/10.1074/jbc.M103114200 JBCHA3 0021-9258 Google Scholar
A. U. KhanM. Kasha,
“Chemiluminescence arising from simultaneous transitions in pairs of singlet oxygen molecules,”
J. Am. Chem. Soc., 92
(11), 3293
–3300
(1970). http://dx.doi.org/10.1021/ja00714a010 JACSAT 0002-7863 Google Scholar
R. Boodaghianset al.,
“Intensities of hot bands in the dimol emissions of singlet molecular oxygen, ,”
J. Chem. Soc. Faraday Trans. 2, 78
(8), 1195
–1209
(1982). http://dx.doi.org/10.1039/f29827801195 JCFTBS 0300-9238 Google Scholar
A. U. Khan,
“Activated oxygen: singlet molecular oxygen and superoxide anion,”
Photochem. Photobiol., 28
(4–5), 615
–626
(1978). http://dx.doi.org/10.1111/php.1978.28.issue-4-5 PHCBAP 0031-8655 Google Scholar
K. Blinovaet al.,
“Mitochondrial NADH fluorescence is enhanced by complex I binding,”
Biochemistry, 47
(36), 9636
–9645
(2008). http://dx.doi.org/10.1021/bi800307y BIORAK 0006-2979 Google Scholar
BiographyIrina Baran is an associate professor of medical biophysics at “Carol Davila” University of Medicine and Pharmacy. She received her PhD in physics from the University of Bucharest. Her research focuses on calcium signaling, leukemia therapy, oxidative stress, mitochondrial metabolism, apoptosis, ultra-weak photo-induced luminescence, computational biology. Researcher Id A-8307-2011. She is an editor for the Journal of Visualized Experiments and Dataset Papers in Physics. In 2005 she was awarded the “Dragomir Hurmuzescu” Prize of the Romanian National Academy. Diana Ionescu is an associate professor of medical biophysics at “Carol Davila” University of Medicine and Pharmacy in Bucharest. Her scientific interests regard the effects of Hofmeister anions and flavonoids on the electrical and mechanical parameters of lipid bilayers. Her current research is focused on the study of molecular and cellular mechanisms of action of flavonoids as potential chemotherapeutic agents in human leukemia. She has published over twenty studies in international scientific journals. Simona Privitera received a degree in physics in 2003, and her PhD in physical engineering, in 2007, at the University of Catania. She is a research fellow at Faculty of Engineering, Catania University, for the activities relating “Modeling aspects and applications of photo-induced ultra-weak luminescence emitted by biological systems,” since 2009 to 2011. She is now a teacher of mathematics and physics in high school. Agata Scordino received the degree in physics in 1980, and now is associate professor of experimental physics with the University of Catania. Principal scientific interests: interaction of electromagnetic waves and rough metal surfaces, spontaneous and photoinduced ultraweak photon emission from living systems, development of innovative instrumentation for photons detection. She has performed research in collaboration with researchers from Italy, Netherlands, Poland, Romania, Switzerland, United Kingdom, and Ukraine. Maria-Magdalena Mocanu received her PhD from the University of Bucharest, Romania, in 2005. She joined the Biophysics Department, “Carol Davila” University of Medicine and Pharmacy in 2008, where she studies: (1) EGF receptor family members and their signaling pathways in cancer; (2) protein misfolding associated with amyloidogenesis. She was trained in the Biophysics and Cell Biology Department, University of Debrecen, Hungary, and in the Max-Planck Unit of Structural and Molecular Biology, Hamburg, Germany. Francesco Musumeci is full professor of applied physics with the University of Catania and visiting professor with University of Kyoto, Japan. His research activities were focused on the field of solar energy, on the interaction between electromagnetic waves and rough metal surfaces, on the dielectric properties of aqueous solutions. His current research interests include the investigation of ultraweak luminescence, spontaneous and photoinduced, from biological systems. Rosaria Grasso received the degree in electronic engineering in 2005 and got her PhD in physical engineering in 2009 at the University of Catania. She has worked on detection of ultra-weak induced photon emission from biological systems and the study, design and testing of detectors for time resolved imaging with single-photon sensitivity. She also investigated the non-thermal effect induced by radio frequency electromagnetic fields on cell cultures. Marisa Gulino got the degree in physics at the University of Catania, Italy, in 1996. In 2000, she got the PhD in physics “cum laude” at the University of Catania, Italy. She has been a postdoc researcher with Katholieke Universiteit Leuven, Leuven, Belgium. Then, she has joined the biophysical research group of the University of Catania, as a researcher. She is now assistant professor with the University “Kore” of Enna, Italy. Iftime Adrian is a lecturer in medical biophysics, with research interests in image data processing, visual field analysis and its digital reconstruction and also some particular fields of optical spectroscopy. He brings expertise in high precision calibration and check-up of research experimental setups. With a PhD in psychophysics, he contributed in the research design of the experiments aiming to minimize the observer bias. Ioana Teodora Tofolean is currently a medical student in the sixth year at “Carol Davila” University of Medicine and Pharmacy in Bucharest. During the high-school period she was a participant and a medalist in several international chemistry olympiads. In 2008 she became a team member of the research projects in the Biophysics Department of the “Carol Davila” University. She is a co-author for four papers published in ISI journals and other scientific reports. Alexandru Garaiman is currently a medical student at the faculty of medicine, “Carol Davila” University of Medicine and Pharmacy in Bucharest, Romania. In 2012/2013 he was enrolled as a student at the University of Leipzig in Germany within the lifelong learning programme. He is a research team member at the Biophysics Department of the “Carol Davila” University since 2010. He is a co-author for two papers published in ISI journals. Alexandru Goicea was born in 1992 in Constanta, Romania. He is a medical student at the faculty of medicine, “Carol Davila” University of Medicine and Pharmacy in Bucharest, Romania. In 2011 he joined the research program at the Biophysics Department of the “Carol Davila” University where he was trained in cell cultures and spectrofluorimetry. His current research activity concerns the antitumoral effects of flavonoids and oxidative stressors in human leukemia cells. Ruxandra Irimia is a medical student at the faculty of medicine, “Carol Davila” University of Medicine and Pharmacy in Bucharest, Romania. In 2010 she joined the research program at the Biophysics Department of the “Carol Davila” University where she was trained in cell cultures, spectrofluorimetry and flow cytometry. She is currently involved in a research project concerning the chemotherapeutic properties of flavonoids in human leukemia cells. Alexandru Dimancea is a medical student at the faculty of medicine, “Carol Davila” University of Medicine and Pharmacy in Bucharest, Romania. In 2011 he joined the research program at the Biophysics Department of the “Carol Davila” University where he was trained in cell cultures and spectrofluorimetry. His current research activity concerns the mitochondrial metabolism and apoptosis, as well as the antitumoral effects of flavonoids and oxidative stressors in human leukemia cells. Constanta Ganea is full professor and head of the Biophysics Department at “Carol Davila” University of Medicine and Pharmacy in Bucharest, Romania. Her research interests cover membrane transport, oxidative stress, and flavonoids, mitochondrial metabolism, cancer therapy. She published over 40 publications in ISI journals including Journal of Biological Chemistry, Biophysical Journal, Langmuir, BBA, Biochemistry (US), Pfluegers Archiv, Researcher Id. A-8561-2011. She coordinated several national and international research projects in the field of biophysics. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||




