|
|
1.IntroductionFluorescence is a two-phase process during which photon absorption is followed by photon emission. Its high sensitivity and specificity1 are harnessed in sensing, imaging, and manipulation. In fluorescent imaging and measurement techniques, the fluorophore molecules of the sample are excited by illumination, then the fluorescent photons emitted by the relaxing molecules are collected and registered. The number of fluorescent photons refers to the number of fluorophore molecules within the excited region. Thus, based on the recorded data, a computer can depict the fluorophore distribution as a microscopic image of the sample. Moreover, the temporal changes and fluctuations of the fluorescent signal carry information about the diffusion of fluorophore molecules into and from the excited volume, which is exploited in fluorescence correlation spectroscopy (FCS)2,3 and fluorescence recovery after photobleaching (FRAP) (also known as fluorescence photobleaching recovery) measurements.4 There are different special techniques to increase the spatial resolution of imaging. In two-photon microscopes, focused light pulses generated by a mode-locked laser excite the fluorophore molecules of the sample.5 At the focal region, the intensity of illumination is high enough to provoke two-photon excitation, during which two photons are absorbed simultaneously while the molecule attains an excited state. However, moving further away from the focus, the probability of two-photon absorption decreases rapidly, because its rate is proportional to the square of the irradiating light intensity (see Sec. 2.2). The fluorescent photons can, therefore, be considered to originate from a small (subfemtoliter6) volume around the focus. The recently worked out super-resolution microscopy techniques circumvent the diffraction limit of the light microscopes formulated by Abbe and Rayleigh. These methods apply for instance special illumination patterns (as in the case of structured illumination microscopy) or special nonlinear excitation (in stimulation emission depletion microscopy), or limit the number of simultaneously emitting molecules to promote their individual localization (in photoactivated localization microscopy, fluorescence photoactivation localization microscopy, and stochastic optical reconstruction microscopy).7,8 In the illuminated sample, not only fluorescence (i.e., “excitation followed by photon emission”) takes place, since the fluorophore molecules can also undergo further possible state transitions: the excited molecule can return to the ground state via internal conversion without photon emission, it can reach the triplet state by intersystem crossing, or it can absorb more photons. These absorbed photons can cause photochemical reactions called photobleaching, which destroys the fluorescent ability of the fluorophore molecule irreversibly. Photobleaching has a significant influence on fluorescence: on one hand, it is a major limit of image quality in two-photon microscopy,9 and it also causes considerable difficulties in FCS;2,3 on the other hand, it is a fundamental process in FRAP measurements. In order to gain accurate information on the sample (e.g., the spatial distribution or the diffusion constant of the fluorophore molecules), one has to know as precisely as possible where the fluorescent photons originate from. In other words, one needs to be aware of the detailed spatial profile of fluorescence. This needs the consideration of the following factors:
In this paper, we present a numerical model which combines all these considerations contrary to previous works that combined some but not all of the mentioned elements. The models described in the preceding publications can be divided into two groups: models in the first group focus on state transitions but without taking into account the spatial profile,9–11 and some of them also lack the consideration of photobleaching.12 Models in the second group deal with the spatial profile; however, they do not distinguish real photophysical states, but only an “active” and a photobleached state (and in one case, also triplet dynamics), which corresponds to the case in which photobleaching affects molecules in the ground state.2,3,13–15 Based on our model, we implemented a simulator program which enables the investigation of the spatial distribution of fluorescence and photobleaching in two-photon excited samples. The model arrangement is as follows: a diffusible, initially homogenous solution of a fluorophore is excited by a focused laser pulse train. The laser light evokes two-photon excitation and other state transitions according to the applied photodynamic model. A substantial difference between the simulations based on the presented model and real experiments is that the simulator uses a continuum model which determines the probabilities and expected values of the events instead of handling the discrete stochastic events (e.g., photon emission and photobleaching) that occur in practice. Moreover, the simulator can reveal the spatial distribution of photon emission within the excitation volume unlike a real two-photon microscope, which counts the photons emitted from the whole excitation region without considering their precise source location. The simulated arrangement can serve as a basic model for several measurement and imaging techniques such as the aforementioned two-photon and super-resolution microscopy, FCS, and FRAP. In Sec. 2, we present our computational model in detail: the model of the illumination, the photodynamic model of the fluorophore, and later on, the diffusion model and the model of the photon detection. We also recall the underlying physical principles. Afterward, in Sec. 3, we present our simulations. Finally, we discuss the results (Sec. 4) and close the paper with conclusions and acknowledgments. 2.Simulation ModelWe modeled the following arrangement: a pulsed laser beam is focused on a homogenous fluorophore solution in which excitation, relaxation processes, photon emission, and photobleaching take place (Fig. 1). The sample is divided into cubic cells among which diffusion occur. The simulator calculates the number of fluorophore molecules in each photodynamic state (see Sec. 2.2.2) and the number of emitted fluorescent photons for every volume cell in each time step. Fig. 1Scheme of the simulation arrangement. The red hour glass shape denotes the illumination laser beam. The dotted rectangular box shows a possible simulation volume instead of which a cylindrical volume is used: the actually simulated volume cells are denoted by the small blue cubes. Axis points toward the direction of light propagation. 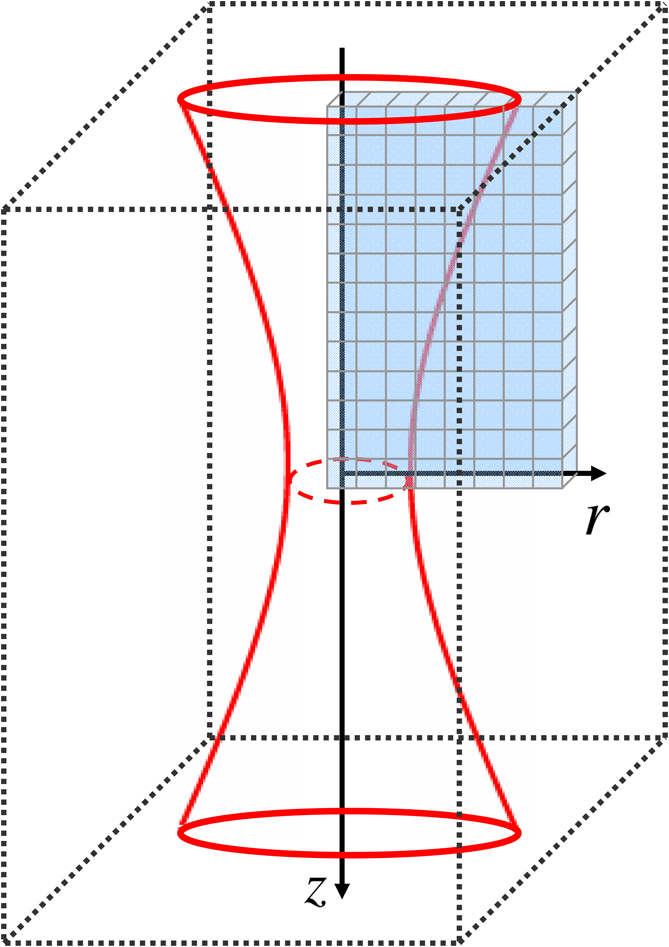 The arrangement has a cylindrical symmetry with respect to the optical axis of the illuminating laser beam. Thus, a volume cell can be referred to by two cylindrical coordinates, namely the radial coordinate denoting the distance from the beam axis and the axial coordinate denoting the distance measured from the focus along the beam axis; the origin of the coordinate system is the focus of the illuminating beam. Taking advantage of the cylindrical symmetry, computational complexity can be remarkably reduced: there is no need to simulate the whole illuminated region (denoted by the dotted rectangular box in Fig. 1), but it is sufficient to simulate only one radial slice of it,2 which reduces the number of volume cells from to . The arrangement is also mirror symmetric with respect to the focal plane , which enables a further reduction of the number of volume cells by a factor of . 2.1.Model of the IlluminationA monochromatic, continuous wave laser beam is commonly described as a Gaussian beam (also called Gaussian–Lorentzian profile,9 which is a solution of the paraxial Helmholtz equation).16 However, to generate a photon flux that is intense enough to provoke two-photon excitation, mode-locked lasers are applied, which emit pulses with a duration in the femtosecond range. For instance, titanium:sapphire lasers are prevalently used in two-photon microscopes. They emit 100-fs-long laser pulses with a wavelength between 700 and 1050 nm,6 which corresponds to about 30 to 40 cycles of the electromagnetic wave. Assuming that the temporal shape of these pulses is a Gaussian function, thus their Fourier transform is also a Gaussian function,17 and we can determine that the full-width at half-maximum (FWHM) bandwidth of such a Gaussian pulse is about 55 THz, which corresponds to 15% of the typical central frequency of 375 THz (800 nm). Consequently, the monochromatic continuous wave Gaussian beam offers an acceptable approximate description for such pulses. In this work, we modeled the illuminating light as a train of focused laser pulses, whose temporal profile is Gaussian (Fig. 2): In which denotes the standard deviation of the Gaussian pulse with FWHM of . is the peak power of the pulses, which can be determined from the long-time-averaged laser power as where is the pulse repetition time (i.e., the inverse of the pulse repetition rate).During the simulations, we consider the laser beam to be monochromatic. (However, as pulses get shorter, the error of this approximation increases, the spectrum of the pulses expands, and the space–time profile of the beam varies significantly. For the description of such pulsed beams, see the work of April.18) In the Gaussian beam, the light intensity at a point is given by the equation where in which is the Rayleigh range, is the waist radius, is the wavelength of the excitation light, and is the beam divergence.16 Subscript refers to excitation for the sake of the distinction from the detection profile (see Sec. 2.4). For a continuous wave Gaussian beam, the light intensity at the focus can be determined from the laser power asWe suppose that this relationship also remains approximately valid for our pulsed case, i.e., we can assume that in Eqs. (4) and (8), the instantaneous intensity at the focus as well as the instantaneous intensity at point follows the laser power without delay. Therefore, the light intensity at a point and time instance can be calculated as moreoverIn our model, we neglect the scattering and the attenuation of light in the sample. By the use of the Gaussian beam, the illumination is described as a scalar field of light intensity, i.e., only the magnitude of the electric field is considered, but not its direction. We assume that the orientation of the fluorophore molecules is uniformly random, thus the influence of the orientation is involved in the absorption cross-section. Furthermore, we assume that the lifetime of the relaxation process coupled to photon emission is much longer than the Brownian rotational lifetime of the fluorophore molecules; therefore, the emitted fluorescent light is considered to be depolarized.19 2.2.Photodynamic Model of the Fluorophore Molecules2.2.1.Theoretical description of the rates of one- and two-photon absorptionsIn the following, we recall the theoretical background of one- and two-photon absorptions according to the books of Boyd20 and Masters and So1 using the semiclassical model for the description of light–matter interaction. In this approach, the matter is quantized (it is built of particles), whereas light is handled as an electromagnetic field. In the absence of radiation, one can describe the atomic system with the Schrödinger equation: whose eigenfunctions and eigenvalues give the possible states and energy levels of the system, where is the state function, is the momentum, is the mass of the particle, is the potential energy at point , is the imaginary unit, and is the reduced Planck constant. According to the electric dipole approximation, illumination of the system can be interpreted as a perturbation during which the electric field induces electric dipoles. Consequently, a new term is to be added to the Schrödinger equation when the atomic system is illuminated: where is a vector connecting the separated charge in the ’th electric dipole induced by electric field of angular frequency at time . Assuming that the illuminating light is monochromatic and linearly polarized, from Eq. (12) applying the first-order time-dependent perturbation theory, one can express the rate of the transition from the ground state to an excited state due to one-photon absorption as where is the one-photon absorption cross-section in units of , and is the light intensity or, more precisely, photon flux, measured in units of . Similarly, in the case of two-photon absorption, the second-order time-dependent perturbation theory results in the following rate for the transition from the ground state to an excited state , via a virtual state : where is the two-photon absorption cross-section in units of . The absorption cross-sections can be measured or they can be determined theoretically by Fermi’s golden rule: and where denotes the angular frequency of the illuminating light, is the refractive index, is the speed of light, and is the density of states. The theoretical derivation of the absorption cross-sections requires the knowledge of the transition moment , which can also be calculated theoretically, namely from the wave functions and of the two states:Nevertheless, for larger molecules, the calculation of the state functions is practically impossible because of the computational complexity of the problem. In our simulations, the cross-sections are to be given as input parameters. Finally, we note that the connection between the light intensity and the electric field is 2.2.2.Photodynamic states and transitions in our modelWe model the states and state transitions by a multilevel photodynamic model taken from the literature9–11 with modifications. The model includes four different photobleaching routes starting from the excited states. It contains the following states (Fig. 3): ground state , excited state , triplet state , and four photobleached states , , , and . We neglect vibrational and rotational states; therefore, we consider the electronic states to be discrete (sharp). In the model, the following transitions are possible:
Fig. 2The Jablonski diagram of the fluorophore represents the photodynamic states and the possible state transitions of the model. , , and denote the ground state, excited state, and triplet state, respectively; , , , and indicate the photobleached states. Solid red arrows pointing upward denote photon-absorption-induced transitions (group 1), arrows pointing downward denote the relaxation transitions (group 2) with (solid blue line) or without photon emission (dashed black lines), and denotes the rate constant of each transition. 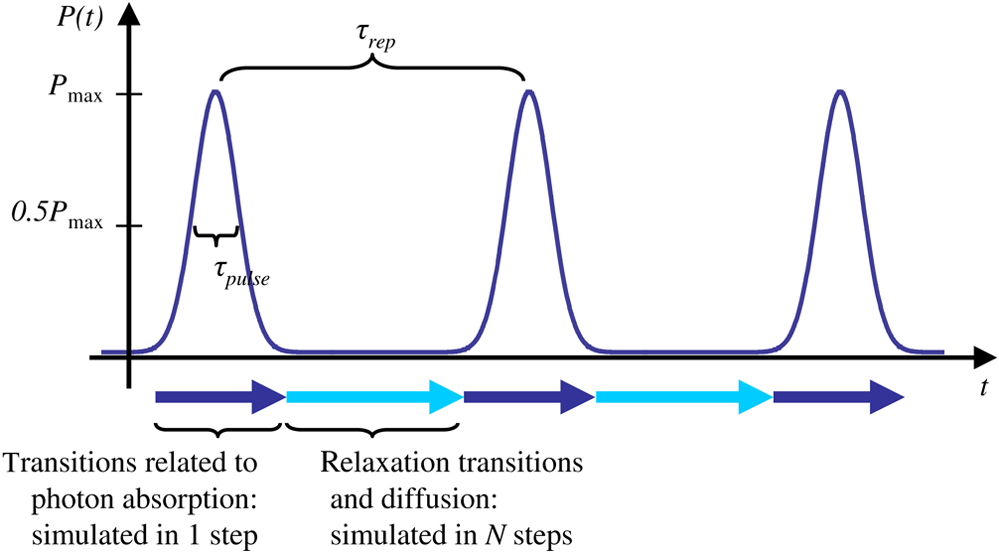 We omit stimulated emission as it is neglected also in several referenced papers.9,11,19 The transition from state to state is described as follows: the time derivative of the number of molecules in state in volume cell at time instant (supposing the change is due only to transition ) is whereas the time derivative of the number of molecules in state (supposing the change is due only to transition ) is where is the rate constant of the transition. If more than one transition affect a state, then the changes of the numbers of molecules deriving from each transition are added up.The photodynamic processes in the model can be divided into two groups:
We suppose that the pulse duration is much shorter than the time constant of the relaxation processes (i.e., , , , ), furthermore the pulse length is much shorter than the pulse repetition time (i.e., ). Therefore, during a light pulse, the relaxation processes can be neglected and it can be assumed that a fluorophore molecule is excited atmost once per pulse.9–11,19 Consequently, it is reasonable to construct two differential equation systems: the first one describes the photon-absorption-induced processes taking place during the light pulses (corresponding to group 1); the second one expresses the relaxation transitions occurring during the interpulse periods (corresponding to group 2).10 Thus, the following two differential equation systems have to be constructed for each volume element: andThe vectors on the right side contain the number of molecules in the given cell in the corresponding states; the vectors on the left are built from the time derivatives of the molecule numbers; whereas the matrices contain the rate constants characterizing the transitions. The molecule numbers and their time derivatives in both equations, and the rate constants in Eq. (23), depend on spatial coordinates and time ; however, for the sake of simplicity, it is not signed here [as opposed to Eqs. (19) and (20)]. On the other hand, rate constants in Eq. (24) are independent of space and time as already mentioned. 2.2.3.Computation of the transition probabilitiesBased on the ordinary differential equation system formulated in Eq. (23), one can calculate the probability of each photon-absorption-induced state transition during one laser pulse for every volume cell. Because the pulse train consists of identical laser pulses, it is enough to calculate these probabilities once. , , and are the three states from which transitions are possible. Accordingly, the simulator solves the differential equation system with three different initial conditions formulating that the molecule starts from state , , or . In our model, the time profile of the illumination intensity is Gaussian; thus, it has an infinite support. Nevertheless, the numerical integration is performed on the arbitrarily chosen domain spanning from to , where denotes the standard deviation of the Gaussian function describing the laser pulse. The relaxation transitions [together with the diffusion (see Sec. 2.3)] taking place between two consecutive laser pulses are simulated in steps; that is, the simulation time step is . Similar to the photon-absorption-induced transitions, the simulator calculates the probability of each relaxation transition occurring between laser pulses by solving Eq. (24) numerically for . The solution is performed with two different initial conditions which describe that the molecule starts from state or . [For Eq. (24), there exists also an analytical solution, namely exponential functions. Transition is a two-step decay, for which Bateman gave a solution.21] The numerical solution of the differential equation systems is performed by the ode45 MATLAB function applying the Dormand-Prince method, which combines explicit fourth and fifth order Runge-Kutta formulae.22 As a result, the simulation of a state transition is simplified to the transfer of a certain fraction of the molecules from one state to another according to the probability calculated initially. Pulse saturation and ground state depletion are inherently taken into consideration by the model. 2.3.Diffusion ModelAs explained above, the sample is modeled as an initially homogenous, diffusible fluorophore solution, which is divided into cubic cells. Hereby, we give the detailed model of the isotropic diffusion taking place in the sample. 2.3.1.Theoretical description of diffusion: Fick’s lawThe transportation of molecules due to the concentration gradient is described by Fick’s law of diffusion: where is the vector of particle flux density, is the diffusion constant, and is the concentration of the particles.23 The relation between the diffusion flux and the concentration is given by the continuity equation, which expresses the conservation of the particles:From Eqs. (25) and (26), the diffusion equation can be obtained: The solution of Eq. (27) is which describes the concentration evolved by diffusion at point at time if the matter is placed at the origin, .23 Separating the diffusion along the three axes, we obtain i.e., the concentration of the molecules along each axis is described by a Gaussian distribution with zero mean and variance of .2.3.2.Discretized diffusion modelSimulation needs discretization both in space and time. The spatial discretization is achieved by the cubic mesh described at the beginning of Sec. 2, whereas the time step of the diffusion simulation is as was stated in Sec. 2.2.3. (In Sec. 3.1.3, we present that even can be a convenient choice.) In every time step, a molecule can stay at the same volume cell or can diffuse to one of the two neighboring cells along each axis; the probability of diffusion is the same toward the negative and positive directions. Let be a discrete random variable which denotes the dislocation of a molecule from the origin along axis . It can take three values:
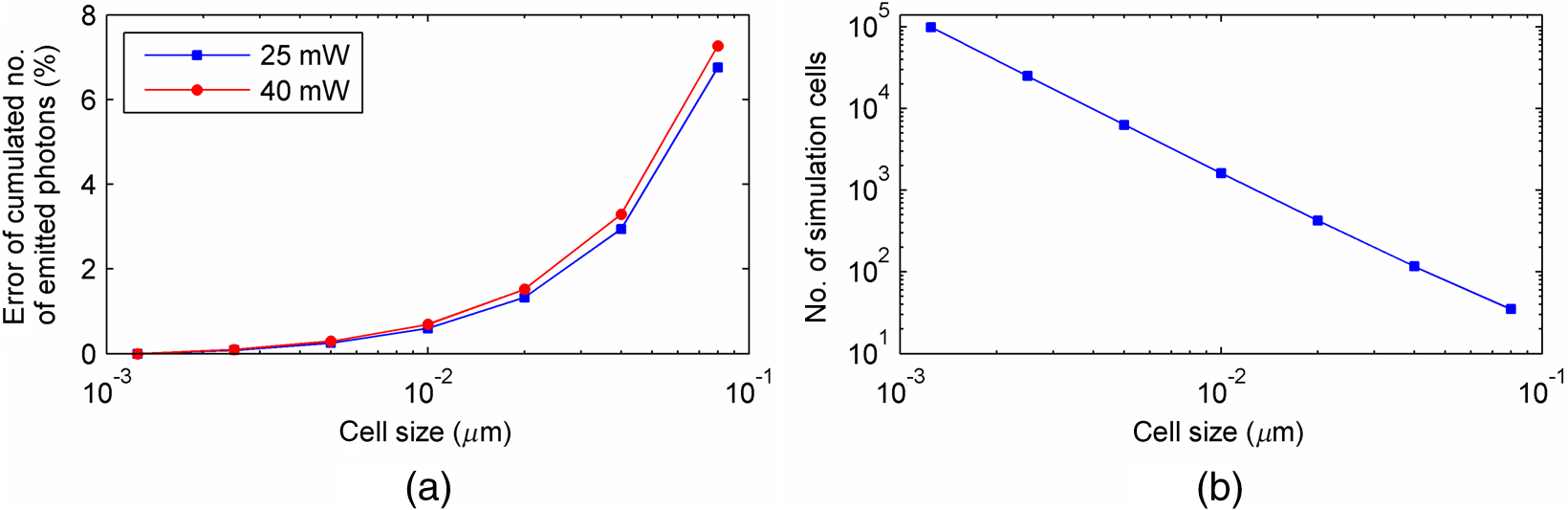
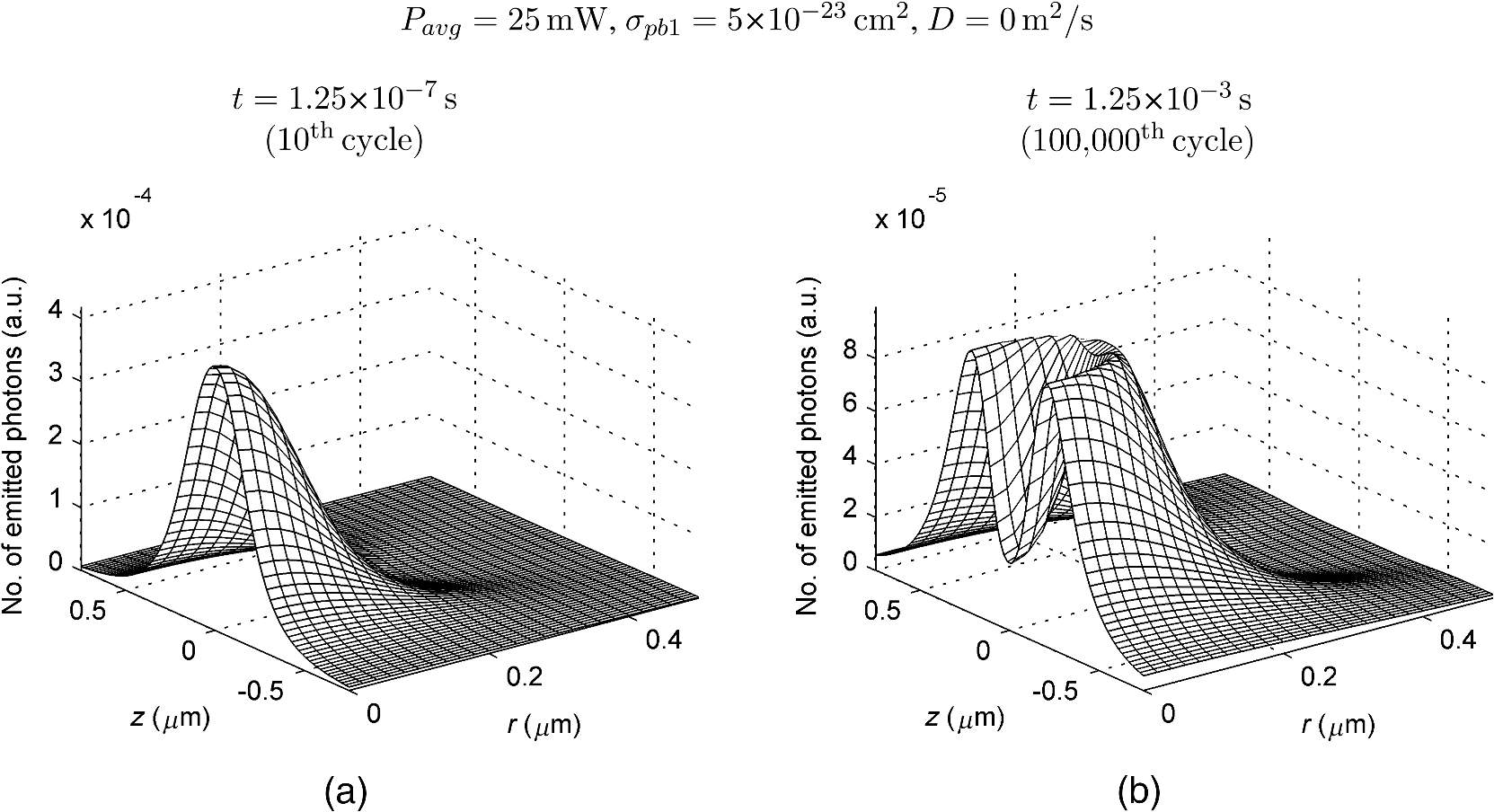
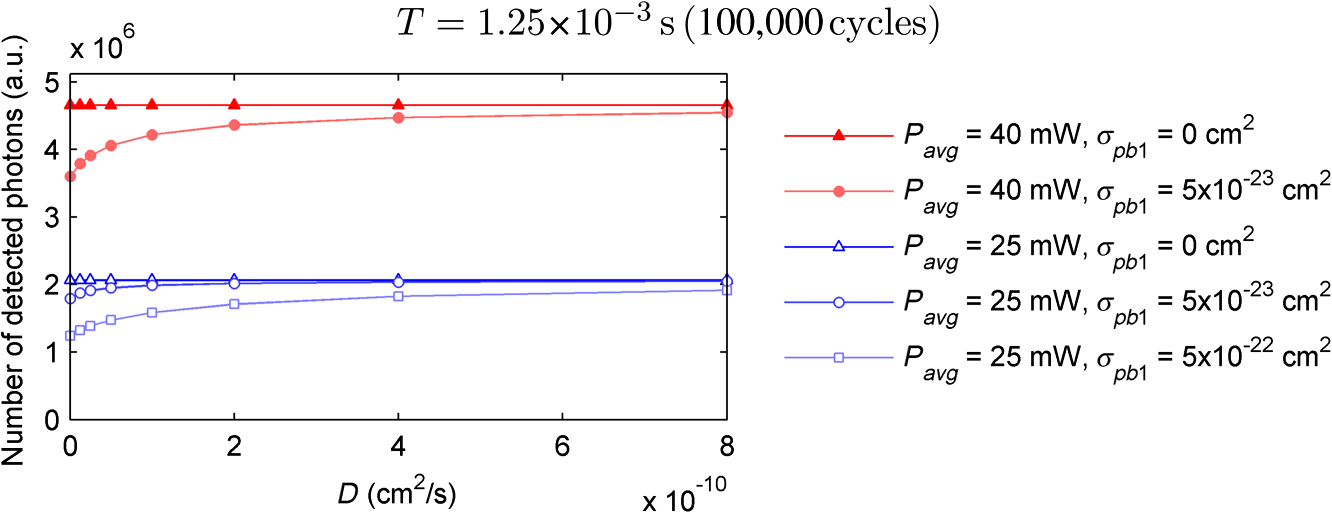
The probabilities that belong to these three events are the following: where . Therefore, the expectation value and variance of are andLet be a random variable which denotes the dislocation of a molecule after time steps, i.e., According to the central limit theorem,17 as , is asymptotically normal (Gaussian) with mean and varianceEquation (29) states that the distribution of the diffusing matter is Gaussian with zero mean and a variance of ; thus, for the variance of , equation should hold. Substituting into Eq. (35), we obtain for the probability that a molecule moves to one of the neighboring volume cells along a coordinate axis during one time step.2.3.3.Cylindrical symmetryThe three axes along which we describe the diffusion are axes , , and a third axis , which is perpendicular to both axes and . Therefore, in order to simulate the diffusion along axis , we need two adjacent volume cell layers lying next to the simulation layer (Fig. 4). Because of the cylindrical symmetry with respect to axis , these adjacent layers are identical. The number of molecules in the cells of these layers is estimated by interpolation based on the values of the simulation layer. For the sake of computational simplicity, linear interpolation is used. Fig. 3The estimation of the neighboring cell layer for the calculation of diffusion is achieved by linear interpolation using the values of the simulation layer. Axis points from the reader into the figure at point . 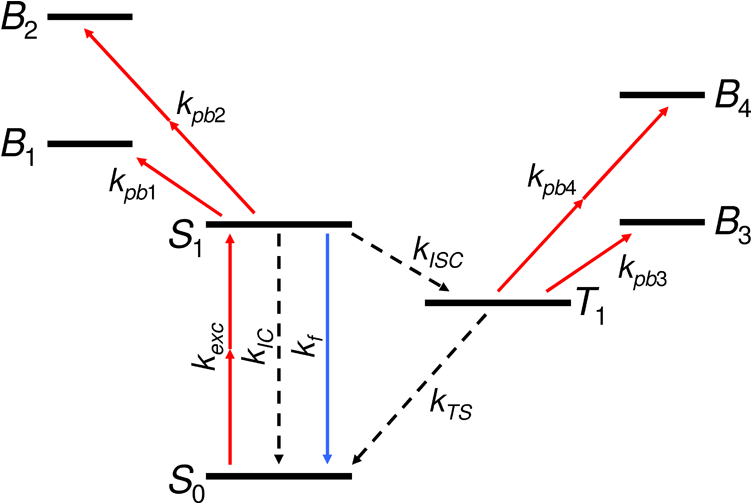 Because of the cylindrical symmetry, the concentration at point has to be equal to the concentration at point since both points are at distance from the origin. From the concentrations and at points and , the concentration at points and is determined by linear interpolation: where denotes the floor function. Because the distance between points and is unity, is equal to the distance between points and .2.3.4.Boundary conditionsDuring the simulation of diffusion, we use zero-flux (Neumann) boundary condition at the outer edges, i.e., the edge layer is repeated and is put beside the simulation region. The cells situated on axes and form “virtual” edges because they are actually in the middle of the simulated region. Therefore, in these cases to preserve the delineated symmetry of the arrangement, not the edge layer (which is located on the axis of symmetry), but the next one is repeated and put beside the simulation region during the simulation. 2.4.Model of the Photon DetectionHere, we present a possible model for photon detection, although in this paper we do not cover the detected number of photons nor the detection profile. The probability that one detects a fluorescent photon originating from the volume element located at is where is the efficiency of the detection, is the fractional solid angle of the observation, and is the observation beam profile.19 In the case of full aperture detection, andIt means that the fluorescent photons are detected with the same probability, independently from the point from where they were emitted. (NA denotes the numerical aperture, and is the refractive index of the sample). However, for confocal observation through a single-mode fiber, and i.e., the observation beam profile is described by a Gaussian beam, whose waist radius is . denotes the wavelength of the emitted fluorescent light which is to be detected. (Subscript refers to detection.).2.5.Process of the SimulationIn the beginning of the simulation, the probability of each state transition is calculated for every volume cell (see Sec. 2.2.3). Then comes the simulation of the state transitions and photon emission evoked by the pulsed illumination. Initially, all the molecules are in the ground state (). At the end of the first light pulse, a fraction of the molecules given by the transition probabilities is transferred to other states by the photon-absorption-induced transitions (group 1 in Sec. 2.2.2). Until the start of the second light pulse, relaxation processes (group 2 in Sect. 2.2.2) and diffusion take place. They are simulated in the same way: some fractions of the molecules determined by the transition probabilities are transferred to other states or to the neighboring cells. These two phases compose one laser cycle and they are repeated for the required times during the simulation process (Fig. 2). The pseudocode of the algorithm is provided in Appendix. Fig. 4The temporal shape of the laser pulse train and the main steps of the simulation. Each laser pulse is described as a Gaussian function with a full-width at half-maximum length of . The time period between two consecutive pulses is . Note that in the figure the ratio of these two periods is distorted for the sake of demonstrativeness; in fact, is much shorter than . Transitions related to photon absorption taking place during the laser pulses are simulated in one step, whereas the relaxation transitions and diffusion occurring between the laser pulses are simulated in (1 or more) steps. 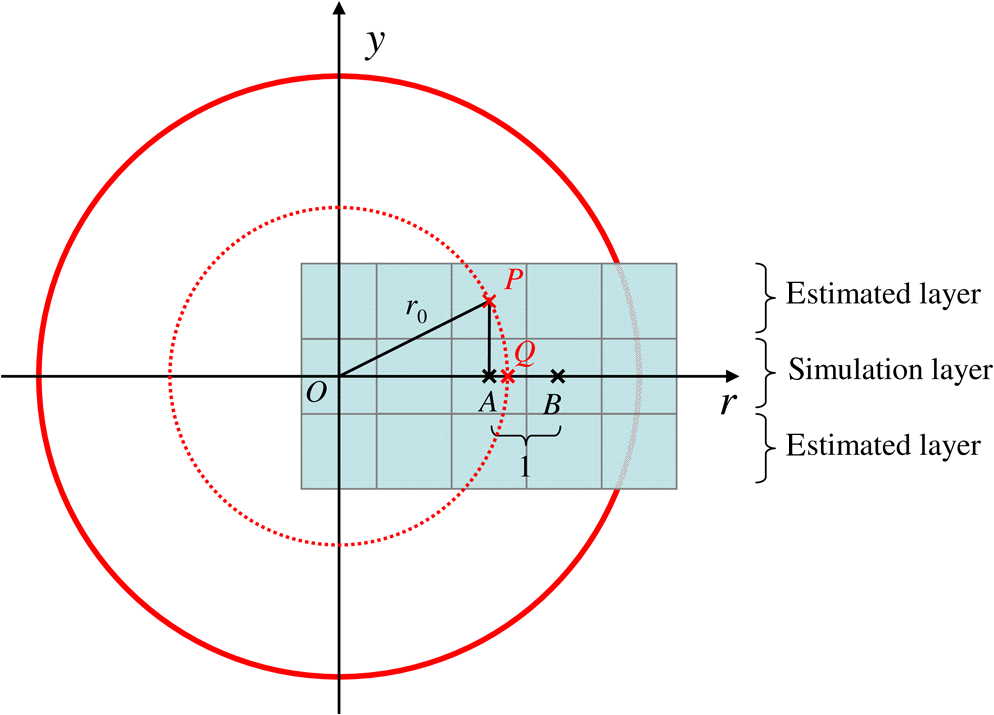 3.Simulations and Results3.1.Selection of the Simulation ParametersOur goal here is, illustrating the capabilities of our model and simulator, to demonstrate by characteristic simulation cases how different factors (laser power, photobleaching cross-section, diffusion coefficient, and intersystem crossing) affect fluorescence. A notable advantage of the simulator is that it makes possible the separate investigation of these factors. We tried to choose the simulation parameters in such a way that they could reflect realistic circumstances. Nevertheless, they do not refer to a given measurement on a real fluorophore by a real device. First, we fixed the following parameters: pulse length , pulse repetition time (i.e., the pulse repetition rate was 80 MHz), and the wavelength of the illuminating light , whose values correspond to the Ti:sapphire laser commonly used in two-photon microscopes. Unless indicated, the diffusion and the relaxation processes are simulated in one step per laser cycle; i.e., and . We set for the beam divergence. In every simulation, we used the arbitrarily chosen for the concentration of the homogenous fluorophore solution; however, this value behaves as a simple multiplier when molecule and photon numbers are calculated by the simulator. We set the fluorophore parameters to be in the same order of magnitude as values found in the literature10,11 for real fluorophores. We used for the excitation two-photon absorption cross-section and 3 ns for the lifetime of fluorescent relaxation. In each presented simulation, we disabled either all the photobleaching routes (by setting their absorption cross-sections to zero) or we enabled one of them (by setting its absorption cross-section to some nonzero value given later). We also disabled internal conversion in all the simulations and intersystem crossing in all but in one virtual experiment setting their lifetimes to and . (The one exception is indicated later, see Sec. 3.2.5.) 3.1.1.Calibration of the laser powerFirst, we ran simulations in order to examine the spatial profile of the excitation probability at different values of the laser power . In this experiment, we disabled photobleaching (i.e., we set , , , and to 0) and diffusion (i.e., we set diffusion constant to 0). We set the radius and height of the simulated cylinder to and , respectively, and the cell size to . We used for the simulation length, which corresponds to 100 laser cycles. The probability of excitation at the focus during one laser pulse increased nearly linearly with the laser power in the case of moderate illumination (5–30 mW) [Fig. 5(a)]. At higher laser powers, the excitation probability saturated, but the number of emitted photons (i.e., the signal) still increased [Fig. 5(b)]. Nevertheless, when we increased the illumination intensity, the fluorescing region expanded [Figs. 5(c) and 5(d)]. It means that the uncertainty of the source location of the fluorescent photons undesirably increased. Based on these results, we chose two laser power levels for the further simulations: 25 and 40 mW. They correspond to the cases when the probabilities that a fluorophore molecule at the focus is excited during one pulse are 70.4% and 95.6%, respectively. Figure 6 depicts the spatial profile of the probability that a fluorophore molecule is excited during one laser pulse in the cases of the mentioned two laser power values. Fig. 5Dependence of (a) the excitation probability at the focus, (b) the number of emitted photons, and (c) and (d) the excitation profile on the illumination laser power. (c) and (d) The probability along axes and that a fluorophore molecule is excited during one laser pulse. The following values were used for the laser power: 5, 10, 15, 20, 25, 30, 35, 40, 50, and 60 mW. The higher the laser power is, the darker and the higher the curve is. Note the difference between the scales of the horizontal axes of (c) and (d). 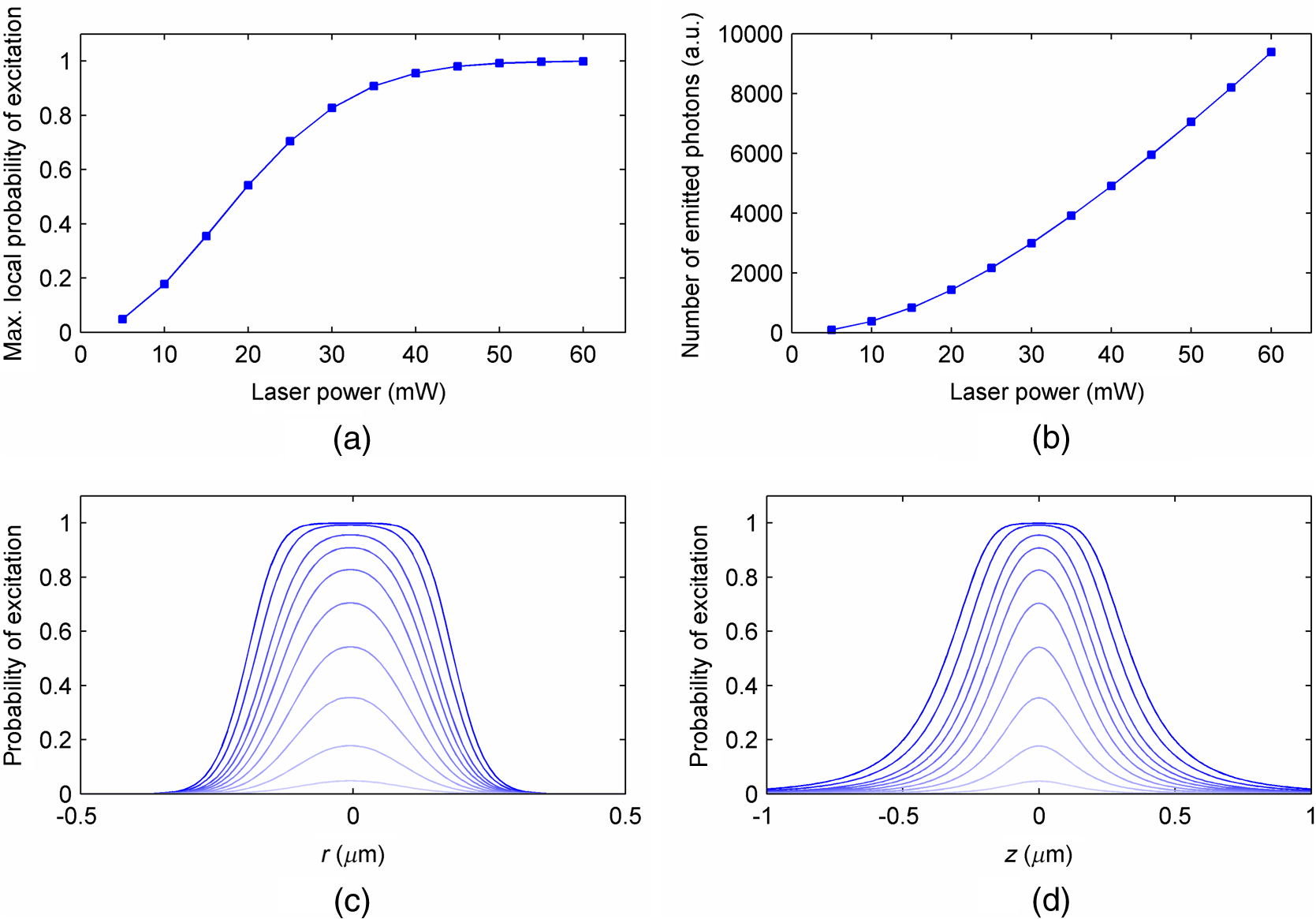 3.1.2.Calibration of the cell sizeIn the next simulation, we checked to what degree the cell size influences the results. Because the illumination intensity changes faster in the space near the focus, whereas its gradient is smaller on the periphery, we ran simulations of length on a small region ( and ) around the focus with different cell sizes. Photobleaching and diffusion were still disabled. Figure 7(a) summarizes the results, whereas Fig. 7(b) depicts the number of simulated cells, which is proportional to the computation time and the memory requirement. Since it was a reasonable trade-off between accuracy and computational time, we retained for the cell size in further simulations. [For this cell size, the error of the emitted-photon number is 0.60% and 0.69% for the two chosen laser power values (25 and 40 mW) considering the case with as the reference.] 3.1.3.Calibration of the simulation of diffusionHere, we investigate how much it modifies the emission profile if the time step of the simulation of diffusion (and relaxation transitions) is reduced. The influence of the diffusion on the fluorescent profile increases with the diffusion constant, the degree of photobleaching, and the length of the simulation. Accordingly, to study the “worst case,” we used and for the laser power and the photobleaching cross-section, respectively, since, among those presented in this paper, this is the setting that results in the highest degree of photobleaching. Furthermore, we set the diffusion constant to the highest value occurring in the presented simulations, namely . The simulation length was (100,000 cycles), and we set the radius and the height of the simulation volume to and , respectively. For , which denotes in how many steps per laser cycle the diffusion and relaxation transitions are simulated, we used eight different values: . In other words, the simulation time step was set to the pulse repetition time of the mode-locked laser and its . Figures 8(a)–8(c) delineate the local relative error of the number of emitted photons per volume cell in the 100,000th cycle in the cases when diffusion and relaxation processes were simulated in 1, 2, and 4 steps per laser cycle. Figure 8(d) shows the maximum local relative error of the number of photons emitted in the 100,000th cycle as a function . In each case, the reference is the photon emission profile obtained for the 128-steps-per-cycle case. These diagrams demonstrate the convergence of the calculated photon emission distribution as the simulation time step tends to zero. Fig. 8(a)–(c) The local relative error of the number of emitted photons per volume cell in the 100,000th cycle in the cases when diffusion and relaxation processes were simulated in 1, 2, and 4 steps per laser cycle. (d) The maximum local relative error of the number of photons emitted in the 100,000th cycle as a function of the number of simulation steps per laser cycle. In each case, the radius and height of the simulation volume were and , respectively, and the reference is the photon emission profile obtained for the 128-steps-per-cycle case. Note the difference between the scales of (a)–(c).  Even in the one-step-per-cycle case [Fig. 8(a)], the relative error was less than 1‰ in the majority of the simulation region. There were two exceptions: some of the cells located at the boundary of the simulation volume (here, the relative error was about 2%) and the outer regions located near the focal plane (where the relative error was about approximately 1%). However, the fact that the rate of excitation (and thus that of photon emission and photobleaching) is small in these regions (see Fig. 6) diminishes the significance of this error. Consequently, even in the presented “worst case” (high photobleaching and intense diffusion), it is enough to simulate the diffusion in one step per laser cycle. Therefore, we used this setting in the following simulations. Now, let us consider the error deriving from the finiteness of the simulation volume. The applied zero-flux boundary condition works properly if the concentration gradients are small at the edge regions. Since the spatial inhomogeneity of the molecules in different states originates from the photophysical transitions taking place mostly at the focus, it can be assumed that increasing the size of the simulation volume decreases the error deriving from the finiteness of the computation region. Thus, to find the appropriate dimensions, we ran simulations in which the radius of the simulation volume was swept from to , while the height of the simulation volume was, in each case, three times larger than the radius. Figures 9(a)–9(c) depict the local relative error of the number of emitted photons per volume cell in the 100,000th cycle in the cases of the three smallest simulation volumes. Figure 9(d) shows the maximum local relative error of the number of photons emitted in the 100,000th cycle within the inner region (of radius and height ) as a function of the radius of the simulation volume. In each case, the reference is the photon emission profile obtained for the largest simulated volume (of radius and height ). These diagrams confirm that as the simulation volume is expanded, the maximum of the local relative error falls rapidly. When the radius and height of the simulation volume are set to and , the error is tolerably small (approximately 2‰ to 3‰); thus, this setting can be regarded as an acceptable trade-off between accuracy and simulation time. If the diffusion constant is smaller, then the degree of the error diminishes to a great extent. It would, therefore, be reasonable to choose a smaller simulation volume in these cases. Nonetheless, for the sake of simplicity and consistency, we set the dimensions of the simulation volume to the aforementioned values ( and ) in all those simulation cases when diffusion is enabled. Fig. 9(a)–(c) The local relative error of the number of emitted photons per volume cell in the 100,000th cycle in the cases when the radius of the simulation volume was , , and , respectively, and the height of the simulation volume was three times its radius. (d) The maximum local relative error of the number of photons emitted in the 100,000th cycle within the inner region (of radius and height ) as a function of the radius of the simulation volume. In each case, the reference is the photon emission profile obtained for the largest simulated volume (of radius and height ). Note the difference between the scales of (a)–(c).  3.2.Investigation of the Three-Dimensional Fluorescent Profile and Its Time Evolution3.2.1.Factors affecting the photon emission profileWe have arrived at the simulations investigating the spatial profile of fluorescence and its changes over time due to photobleaching. Figure 10 depicts 12 simulation cases in which we set different input parameters. We used two different values for laser power (25 and 40 mW), diffusion constant (zero and ), and for photobleaching cross-section : (a value found in the paper of Niesner et al.10) and a value 10 times larger () to simulate increased photobleaching. (The three other photobleaching routes were disabled, i.e., their absorption cross-sections were set to 0.) The heat maps show the normalized number of emitted photons per volume cell during the 10th, 10,000th, and 100,000th laser cycles: in each row, the maximal local value of the fluorescent photon number corresponds to 1. Fig. 10Spatial distribution of photon emission. The images depict the normalized number of emitted photons per volume cell during the 10th, 10,000th, and 100,000th laser cycles. The normalization was carried out row by row: in each row, the maximal local value of the photon number corresponds to 1. In the middle row [(e)–(h)], both the laser power and the photobleaching cross-section were moderate. Even in this case, the decline of fluorescence due to photobleaching was perceptible after an illumination of 10,000 laser cycles (f), and it became severe after 100,000 cycles (g), especially at the focus, where a “dark hole” evolved. Laser power was increased in the first row [(a)–(d)] and the photobleaching cross-section in the third one [(i)–(l)]. In both cases, photobleaching was accelerated. In columns 1–3, diffusion was disabled; however, it occurred in column 4 compensating photobleaching more or less. Note that in the simulations that are presented in column 4 and in the case for which diffusion was enabled, the radius and height of the simulated region were and , respectively; however, only the focal part of the profile is shown here.  Even when both the laser power and photobleaching cross-section were moderate (25 mW and , middle row of the table), the decline of fluorescence due to photobleaching was perceptible after a -long illumination [10,000 laser cycles, Fig. 10(f)], and it became severe after [100,000 cycles, Fig. 10(g)], especially at the focus, where a “dark hole” evolved. When the photobleaching cross-section was set to 10 times higher (third row of the table), then after 10,000 cycles, we got a profile [Fig. 10(j)] which is similar to the one obtained in the case of moderate photobleaching cross-section after 100,000 cycles [Fig. 10(g)]. Not surprisingly, the increase of the laser power also accelerated photobleaching (first row of the table). Diffusion, however, compensated photobleaching more or less (compare columns 3 and 4). (This phenomenon is the foundation of FRAP.) Figure 11 shows the emission profiles depicted in Figs. 10(e) and 10(g) in a three-dimensional plot. 3.2.2.Time evolution of the fluorescent profile in the cases of one- and two-photon-induced photobleachingIn the following simulations, we investigated the time evolution of the spatial profile of fluorescence in the cases of one- and two-photon-induced photobleaching occurring via state . For the former transition, we used again, whereas we set to in order that the probability that a fluorophore molecule being in state and located at the focus is photobleached during one laser pulse would be approximately the same for the two cases. In these simulations, we set the laser power to and disabled the diffusion. The results are summarized in Fig. 12. The first row of the table [Figs. 12(a)–12(d)] depicts the number of emitted photons along axes and during the 1st, 2500th, 5000th, 7500th, 10,000th, 20,000th, 40,000th, and 100,000th laser cycles. The darker the color of the curve, the larger the number of the preceding laser cycles. As more and more laser pulses hit the sample, the number of fluorescent photons falls: we can observe the evolution of the “dark hole” at the focus [see also Figs. 10(c), 10(g), 10(j), and 10(k)]. Another noticeable feature is the appearance of the small “hills” in the fluorescence profile next to the focus. Their width is much smaller than that of the initial fluorescent profile depicted by the uppermost grayish curve in each plot. A distinct difference between the one- and two-photon cases is that two-photon-induced photobleaching is restricted to a smaller volume; therefore, the “walls of the hole” are steeper in this case. Fig. 12Spatial distribution of fluorescence and its time evolution caused by one- and two-photon-induced photobleaching. (a)–(d) The number of emitted photons along axes and during the 1st, 2500th, 5000th, 7500th, 10,000th, 20,000th, 40,000th, and 100,000th laser cycles. The darker the color of the curve, the latter the state is. (e) and (f) The number of emitted photons as a function of the distance of the photon source from the focus. (g) and (h) The characteristic distances of fluorescence over time: , , and denote the minimal radii of spheres which are located at the focus and contain the volume cells that emit 50%, 75%, and 90% of the fluorescent photons within the sphere of radius , located at the focus. The average distance of the location of photon emission from the focus is denoted by linked black squares. Note the difference between the scales of the horizontal axes of (a) and (b), as well as (c) and (d).  Figures 12(e) and 12(f) show the number of emitted photons as a function of the distance of the photon source from the focus. Figures 12(g) and 12(h) show the characteristic distances in the case of different pulse numbers (i.e., different illumination durations): , , and denote the minimal radii of spheres, which are located at the focus and contain the volume cells that emit 50%, 75%, and 90% of the fluorescent photons within the sphere of radius , located at the focus. Along with the average distance of the location of the photon emission from the focus (), these values can be used to characterize the spatial resolution of the fluorescent method. The trends mentioned in the previous paragraph can also be observed in these diagrams: the number of fluorescent photons (i.e., the signal) decreased gradually in time, especially in the focal region. The average distance of the photon emission from the focus shifted toward the periphery and the characteristic distances increased; therefore, the resolution deteriorated because of photobleaching. This effect was more expressed in the case of the one-photon-induced photobleaching. 3.2.3.Effect of the photobleaching cross-sectionNext, we investigated the dependence of the fluorescent photon number on the photobleaching absorption cross-sections. We separately examined the cases of one- and two-photon-induced photobleaching. We used and as references, and we swept these parameters in the range of three orders of magnitude. The results are summarized in Fig. 13. Fig. 13Dependence of the number of emitted photons on the absorption cross-sections that describe photobleaching. The photon numbers are depicted relatively to the reference photon numbers obtained by simulations in which the photobleaching cross-section was set to (a) and (b) (denoted by larger squares). (The radius and height of the simulated region were and , respectively.)  When we decreased by a factor of 32, the number of emitted photons increased by only 28%, whereas when we increased by the same factor, the number of emitted photons fell to 50% of the reference. We obtained similar results for the case of two-photon-induced photobleaching. In this latter case, the emitted-photon number changed less (namely in the range between and with respect to the reference) compared with the one-photon case, which can be explained by the fact that the two-photon-induced photobleaching is more confined in space than the one-photon-induced one (see Sec. 3.2.2). Figure 14 shows the decay of photon emission in time due to photobleaching. The thick curves belong to the aforementioned reference cases ( and ); the reference photobleaching cross-section values were both doubled (darker curves) and halved (lighter curves) five times. Fig. 14Time evolution of the emitted-photon number in the case of different photobleaching cross-sections. The thick curve in each diagram belongs to the simulation case in which the photobleaching cross-section was set to (a) and (b) . These values were both doubled and halved five times: the higher the photobleaching cross-section is, the lower and darker the curve is. (The radius and height of the simulated region were and , respectively.) 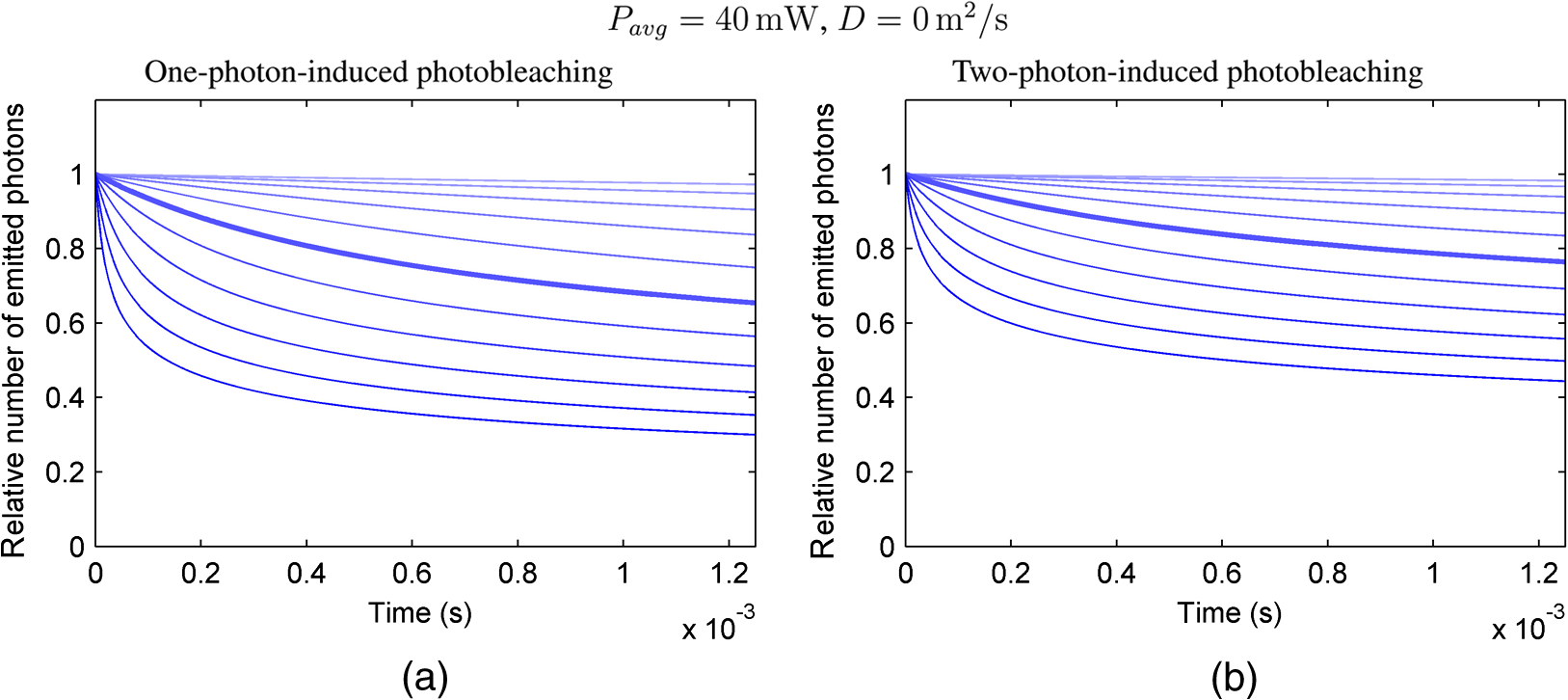 3.2.4.Effect of the diffusionTo investigate the effect of diffusion on the number of fluorescent photons, we swept the diffusion constant from 0 to . (For the sake of comparison, the diffusion constant of glycine in water is about .24) Figure 15 shows the dependence of the number of emitted photons on the diffusion constant. The curves with full and empty triangle marks show the photon numbers obtained for the cases when photobleaching did not occur in the sample; that is, these constant curves indicate the maximal photon number for the given illuminations. We used two different laser powers: 25 and 40 mW. In the 25-mW case, if the photobleaching cross-section was moderate (), diffusion was able to compensate photobleaching. But when photobleaching was more intense (in the case of higher laser power and/or higher photobleaching absorption cross-section), then the number of fluorescent photons decreased even if the diffusion coefficient was large. Additionally, Fig. 15 illustrates that the number of fluorescent photons is more sensitive to the change of the diffusion constant if photobleaching is intensified. 3.2.5.Effect of the intersystem crossingIn the simulations presented up to this point, we disabled intersystem crossing by setting its lifetime to . Here, we discuss the effect of the opening of this relaxation route. Intersystem crossing means a roundabout way for fluorophore molecules to return from the excited state to the ground state, namely without photon emission; thus, it decreases the quantum efficiency. The simulation results (Fig. 16) corresponded to the expectations. The smaller the lifetime of intersystem crossing () is, the fewer photons were emitted, especially if transition was slow, i.e., was large. The return from the triplet state to the ground state takes time, hence with every laser pulse, the number of fluorophore molecules in the triplet state increased toward an equilibrium value [Fig. 17(c)]. Meanwhile, also approaching an equilibrium, the number of emitted photons fell, particularly at the focus [Figs. 17(a) and 17(b)]. As a result, the spatial profile of photon emission became flattened. Fig. 16Dependence of the number of emitted photons (a) on the lifetime of intersystem crossing () and (b) on the lifetime of the transition (). (The radius and height of the simulated region were and , respectively.)  Fig. 17Time evolution of fluorescence in the occurrence of intersystem crossing. (a) and (b) The number of emitted photons along axes and during the laser cycles. The later moment, the darker the color of the curve. (c) The number of emitted photons per laser cycle and the number of molecules in state over time. (The radius and height of the simulated region were and , respectively.) 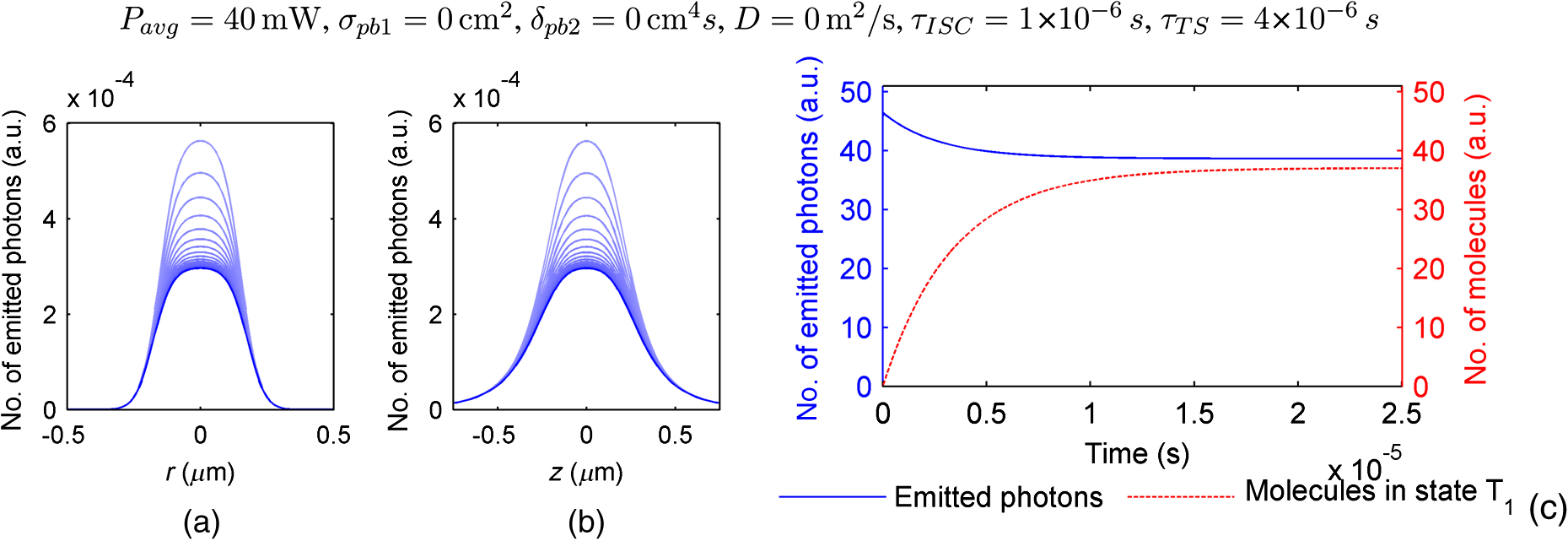 If intersystem crossing is permitted, then the fluorophore molecules in the triplet state might be photobleached by the absorption of one or two additional photons. According to the simulations of this scenario (not presented in detail), these photobleaching transitions affect the photon emission profile similar to those that start from the excited singlet state (see Figs. 10 to 12). Nevertheless, in the former case, the evolution of the photon distribution is also driven by intersystem crossing, which flattens the profile with the “dark hole” at the focus, formed by photobleaching. 4.DiscussionAlthough our model is founded on the physical principles presented in Sec. 2, its validation and calibration require experimental measurements. Besides the approximations and the neglected factors indicated in the description of the model, the most remarkable difference between the presented simulations and real experiments is that our model computes probabilities and expectation values of photon numbers while in reality there are discrete, stochastic events: only whole molecules exist and only whole photons are emitted. Thus, the experimentally measured signals contain fluctuations and noise both in time and space, contrary to the simulation results which are “smooth.” Theoretically, if the experiments are repeated many times, then the average of their results should approach to the simulation results. We implemented the simulator program in MATLAB and ran the simulations on a computer with a 2.4-GHz dual-core processor and 4-GB RAM. The run-time of a simulation case depends on the number of volume cells (which is the function of the cell size and the dimensions of the simulated volume), the number of simulated laser pulses, the number of simulation steps per laser cycle, the number of the enabled state transitions, and whether diffusion is enabled. In terms of these factors, our simulation cases varied from each other to a great extent, which resulted in large differences in their run-times. For instance, the emission profiles in Fig. 10 were calculated within less than a minute if the diffusion was not enabled and the dimensions of the simulation volume were and , but it took 16 min when diffusion was enabled and both the radius and the height of the simulation volume were three times larger. Not surprisingly, increasing the number of simulation steps per laser cycle lengthened the simulation extremely: the 128-steps-per-cycle case resulted in a simulation time of 2.5 h. Presumably, parallelization on a multicore architecture could significantly accelerate the computation, especially when diffusion is disabled in the simulation. There are several possible application fields of the model: taking into account the effects described above and using more accurate fluorescent profiles might facilitate the interpretation of the experimental data of several fluorescent techniques. The model might help to determine the diffusion constants in FRAP experiments and to solve the inverse problems of two-photon and super-resolution microscopy (i.e., the reconstruction of the fluorophore distribution from the measurement data) more effectively, achieving a higher image quality. In addition, the model might promote the optimization of the operating parameters (e.g., illumination laser power) of the measurement device in a particular experiment. The photophysical model and its parameters are, however, unknown for many fluorophores, which can limit the applicability of the model for such purposes. Possibly, the simulator can also assist in the determination of these parameters. In this work, we simulated illumination with a fixed focus; nevertheless, in many fluorescent techniques, the sample is scanned with the laser beam or special illumination patterns are used. Adapting the computational model to such experimental arrangements in order to simulate particular techniques needs further work. In these cases, the cylindrical symmetry vanishes; i.e., the computational time and memory requirements increase to a great extent. The presented model could be easily adapted for the investigation of fluorescence in one-photon-excited samples. However, in this case, as one-photon excitation is less confined in space than two-photon excitation, the size of the simulated volume should be expanded, which would increase the simulation time and the memory requirement. We have already reported simulation results in this field in a conference paper.25 At that stage of our work, we used an equation given by Wang et al.26 to describe the instantaneous illumination light intensity at a given point in a pulsed Gaussian beam. The main drawback of Wang’s equation is that for large values of the radial coordinate , the function is unbounded, which implies that the beam would carry infinite energy.18 This effect becomes more severe as the pulse length decreases. On the other hand, in the case of longer (such as 100-fs-long) pulses, Wang’s equation approaches the continuous wave Gaussian beam. As the function which describes the continuous Gaussian beam is computationally simpler, we finally decided to use this latter one in the simulator. Since the publication of our conference paper,25 we have discovered an error in the former program code: a multiplier factor was missing in the calculation of the illumination intensity from the laser power. This is why we obtained approximately the same excitation probabilities there for laser powers of 30 and 50 mW as here for 25 and 40 mW. 5.ConclusionsWe presented simulation examples which demonstrate quantitatively how the illumination laser power, diffusion, and the photodynamic parameters of the fluorophore (absorption cross-section of photobleaching transitions, lifetime of intersystem crossing, and transition ) affect the number of fluorescent photons and the three-dimensional distribution of fluorescence. We demonstrated how photobleaching changes the spatial distribution of photon emission, producing a “dark hole” in the focus: the simulation results revealed that as more and more laser pulses hit the sample and photobleaching progresses, the most photons are not emitted from the focus but from an ellipsoid situated around the focus. Increased laser power, higher photobleaching cross-section, as well as longer illumination can produce this “dark hole.” On the other hand, diffusion can inhibit this phenomenon. Our results evidenced that photobleaching not only decreases the number of emitted photons, but also deteriorates the resolution of the fluorescent techniques. AcknowledgmentsWe, the authors, gratefully acknowledge the grant from TÁMOP-4.2.1.B-11/2/KMR-2011-0002. We are grateful to the Multidisciplinary Doctoral School of Sciences and Technology, Pázmány Péter Catholic University, Budapest, Hungary and its leaders, Professor Tamás Roska and Professor Péter Szolgay for providing the conditions of the research work. We thank also our colleague, Ádám Fekete for the inspiring discussions. AppendicesAppendix:Pseudocode of the Algorithm
ReferencesB. R. MastersP. T. C. So,
“Classical and quantum theory of one-photon and multiphoton fluorescence spectroscopy,”
Handbook of Biomedical Nonlinear Optical Microscopy, 91
–152 Oxford University Press, New York
(2008). Google Scholar
A. Delonet al.,
“Photobleaching, mobility, and compartmentalisation: inferences in fluorescence correlation spectroscopy,”
J. Fluoresc., 14
(3), 255
–267
(2004). http://dx.doi.org/10.1023/B:JOFL.0000024557.73246.f9 JOFLEN 1053-0509 Google Scholar
Z. PetrášekP. Schwille,
“Photobleaching in two-photon scanning fluorescence correlation spectroscopy,”
ChemPhysChem, 9 147
–158
(2008). http://dx.doi.org/10.1002/(ISSN)1439-7641 CPCHFT 1439-4235 Google Scholar
P.-C. Cheng,
“The contrast formation in optical microscopy,”
Handbook of Biological Confocal Microscopy, 162
–206 3rd ed.Springer, New York
(2006). Google Scholar
W. DenkJ. H. StricklerW. W. Webb,
“Two-photon laser scanning fluorescence microscopy,”
Science, 248 73
–76
(1990). http://dx.doi.org/10.1126/science.2321027 SCIEAS 0036-8075 Google Scholar
W. DenkD. W. PistonW. W. Webb,
“Multi-photon molecular excitation laser-scanning microscopy,”
Handbook of Biological Confocal Microscopy, 535
–549 3rd ed.Springer, New York
(2006). Google Scholar
L. SchermellehR. HeintzmannH. Leonhardt,
“A guide to super-resolution fluorescence microscopy,”
J. Cell Biol., 190 165
–175
(2010). http://dx.doi.org/10.1083/jcb.201002018 JCLBA3 0021-9525 Google Scholar
B. HuangM. BatesX. Zhuang,
“Super-resolution fluorescence microscopy,”
Annu. Rev. Biochem., 78 993
–1016
(2009). http://dx.doi.org/10.1146/annurev.biochem.77.061906.092014 ARBOAW 0066-4154 Google Scholar
J. Mertz,
“Molecular photodynamics involved in multi-photon excitation fluorescence microscopy,”
Eur. Phys. J. D, 3
(1), 53
–66
(1998). http://dx.doi.org/10.1007/s100530050148 EPJDF6 1434-6060 Google Scholar
R. NiesnerW. RothK.-H. Gericke,
“Photophysical aspects of single-molecule detection by two-photon excitation with consideration of sequential pulsed illumination,”
ChemPhysChem, 5 678
–687
(2004). http://dx.doi.org/10.1002/(ISSN)1439-7641 CPCHFT 1439-4235 Google Scholar
C. EggelingA. VolkmerC. A. M. Seidel,
“Molecular photobleaching kinetics of rhodamine 6G by one- and two-photon induced confocal fluorescence microscopy,”
ChemPhysChem, 6 791
–804
(2005). http://dx.doi.org/10.1002/(ISSN)1439-7641 CPCHFT 1439-4235 Google Scholar
K. Paloet al.,
“Calculation of photon-count number distributions via master equations,”
Biophys. J., 90 2179
–2191
(2006). http://dx.doi.org/10.1529/biophysj.105.066084 BIOJAU 0006-3495 Google Scholar
D. Mazzaet al.,
“Role of three-dimensional bleach distribution in confocal and two-photon fluorescence recovery after photobleaching experiments,”
Appl. Opt., 46
(30), 7401
–7411
(2007). http://dx.doi.org/10.1364/AO.46.007401 APOPAI 0003-6935 Google Scholar
N. Smisdomet al.,
“Fluorescence recovery after photobleaching on the confocal laser-scanning microscope: generalized model without restriction on the size of the photobleached disk,”
J. Biomed. Opt., 16
(4), 046021
(2011). http://dx.doi.org/10.1117/1.3569620 JBOPFO 1083-3668 Google Scholar
E. A. Schnellet al.,
“Diffusion measured by fluorescence recovery after photobleaching based on multiphoton excitation laser scanning microscopy,”
J. Biomed. Opt., 13
(6), 064037
(2008). http://dx.doi.org/10.1117/1.3042274 JBOPFO 1083-3668 Google Scholar
B. E. A. SalehM. C. Teich, Fundamentals of Photonics, 2nd ed.John Wiley & Sons, Hoboken, New Jersey
(2007). Google Scholar
G. A. KornT. M. Korn, Mathematical Handbook for Scientists and Engineers: Definitions, Theorems, and Formulas for Reference and Review, Dover Publications, Mineola, New York
(2000). Google Scholar
A. April,
“Ultrashort, strongly focused laser pulses in free space,”
Coherence and Ultrashort Pulse Laser Emission, 355
–382 InTech(2010). Google Scholar
M. Kauertet al.,
“Absolute measurement of molecular two-photon absorption cross-sections using a fluorescence saturation technique,”
Opt. Express, 14 8434
–8447
(2006). http://dx.doi.org/10.1364/OE.14.008434 OPEXFF 1094-4087 Google Scholar
R. W. Boyd, Nonlinear Optics, 3rd ed.Academic Press, San Diego
(2008). Google Scholar
H. Bateman,
“Solution of a system of differential equations occurring in the theory of radioactive transformations,”
Proc. Cambridge Philos. Soc., 15 423
–427
(1910). PCPSA4 0008-1981 Google Scholar
“ode45: Solve nonstiff differential equations; medium order method,”
(2014) http://www.mathworks.com/help/matlab/ref/ode45.html November ). 2014). Google Scholar
Handbook of Physics, Springer, New York
(2002). Google Scholar
R. G. Mortimer, Physical Chemistry, 2nd ed.Academic Press, Burlington, Massachusetts
(2000). Google Scholar
I. B. JuhaszA. I. Csurgay,
“Simulation-based investigation of the three-dimensional distribution of fluorescence and photobleaching in multi-photon excited samples,”
Proc. SPIE, 9131 91310S
(2014). http://dx.doi.org/10.1117/12.2052243 PSISDG 0277-786X Google Scholar
Z. Wanget al.,
“Space-time profiles of an ultrashort pulsed gaussian beam,”
IEEE J. Quantum Electron., 33
(4), 566
–573
(1997). http://dx.doi.org/10.1109/3.563385 IEJQA7 0018-9197 Google Scholar
BiographyImre B. Juhász is a PhD student at the Faculty of Information Technology and Bionics, Pázmány Péter Catholic University, where he received his MSc degree in computer engineering in 2012. His main interest is modeling and simulation of light–matter interaction. Árpád I. Csurgay received his Dipl-Eng degree in 1959, his PhD degree in 1964 from TU Budapest, and his DSc degree in 1973 from the Hungarian Academy of Sciences. He was affiliated with TU Budapest and University of Notre Dame. Currently, he is a professor of applied physics at the PPCU Budapest. He is a fellow of the IEEE and a member of the Hungarian Academy of Sciences and Academia Europaea. |