
|
|
1.IntroductionLeft ventricular remodeling (LVR) as a consequence of acute myocardial infarction (AMI) appears on both macroscopic and microscopic levels. Changes with regard to the macroscopic level refer to the modification of shape, size, and wall thickness of the myocardium. The disproportionate thinning and dilatation of acutely infarcted myocardium is caused by extensive deposition of collagen as well as an increase in the tensile strength. In 1978, Hutchins and Bulkley1 described this process and it was termed “infarct expansion.”2,3 It led to microscopic changes related to a reduction in the number of cardiomyocytes and alterations in connective tissue involving the degradation of connective compartments and proliferation of fibroblasts in the cardiac muscle. However, the myocardium is more than myocytes; it is also an extensive network of extracellular matrix (ECM) which contains collagens and fibrillar proteins, the structural support to resistant cells. The ECM provides a viscoelastic scaffold consisting of type I and III collagen.4,5 The remodeling of the ECM plays a major role in the changes that take place in the LVR.6–8 Whittaker et al.9 demonstrated the significant damage of the myocardial collagen network as early as 1 day after infarction. Furthermore, the authors reported that fibroblasts are responsible for creating the microstructure because of the thickness and crosslinking of collagen fiber bundles, which explains the properties of infarct scar tissue.10,11 Therefore, cardiac wound repair after myocardial infarction involves phases which include an inflammatory phase and tissue remodeling phase. The first phase starts early after coronary artery occlusion and involves degradation of normal ECM, invasion of inflammatory cells at the site of the injury, and the induction of bioactive peptides and cytokines. Degradation of the ECM during the acute phase is an essential event which is related to the appearance of inflammatory cells and the proliferation and maturation of macrophages and fibroblasts. Furthermore, it also provides the necessary substructure for scar formation.12 The concentration of collagen degradation products (CDPs) may reflect the process of LVR. The specific structure of collagen provides its resistance to proteolytic degradation except by matrix metalloproteinases (MMPs). The activated MMPs require the proteolytic cleavage of a propeptide sequence. The classes of MMPs that may have particular relevance to myocardial remodeling possessing high substrate specificity for fibrillar collagens are MMP1, MMP8, and MMP13 and other ECM proteins such as proteoglycans, versican, perlican, and aggrecan.5,13 MMPs are capable of hydrolysing type I, II, and III collagens at the Gly775–Leu776 or Gly775–Ile776 peptide bond of the corresponding α chains.14 Type I and type III collagens are the main fibrillary constituents in developing tissue.15 They are characterized by their ability to assemble into highly orientated supramolecular aggregates with a characteristic suprastructure and the specific quarter-staggered fibril-array with diameters between 25 and 400 nm.16,17 Type III collagen is a homotrimer of three -chains and it is widely distributed in collagen type I containing soft tissues.16 It is a significant component of reticular fibers in the interstitial tissue, including vessels.18 Type III collagen has slightly different functions compared to type I collagen. Type I collagen confers tensile strength and resistance to stretch and deformation, whereas type III collagen fibers confer resilience.19 The cross-linked N-terminal telopeptide (IIINTP) is one of the products from collagen catabolism which is assessed in tissue degradation products as a marker of mature type III collagen.20 Optical methods provide a possibility to study biological samples quickly and often noninvasively. Time-resolved fluorescence spectroscopy (TRFS) is a powerful tool in physics and chemistry21 which can be successfully applied in medical diagnosis. Herein, the fluorescence of a sample is measured as an approximately exponential (often multiexponential) decay function of time as a result of the radiative relaxation of the molecules after a remarkably short pulse of excitation light. Many biological macromolecules, including CDPs, which exhibit complex intensity decay patterns, could be distinguished by different fluorescence lifetimes (FLTs). The solution of this complexity is the picosecond time resolution obtained with time-correlated single-photon counting (TCSPC). It is possible to extend the data collection by multiple excitation/emission cycles, even if a sample consists of just a few molecules.22,23 The aim of this study was to evaluate the potential diagnostic usefulness of TRFS in the assessment of type III CDPs. It has been done using the in vitro model of CDPs and human plasma by checking the influence of different amounts of CDPs on the FLT of plasma. This preliminary experiment was designed to establish if CDPs’ characteristics by FLT might be visible under determined conditions. 2.Materials and Methods2.1.Collagen HydrolysisPurified type III collagen from human placenta (Sigma-Aldrich C-4407) was cleaved with MMP-1 human (Sigma-Aldrich SRP3117). Type III collagen was dissolved in MMP buffer (100 mM Tris HCl, 100 mM NaCl, 10 mM , 2 mM Zn acetate; pH 8.0). MMP cleavage was performed by mixing of type III collagen and of MMP-1 in 1 mL of MMP buffer. As a control, of collagen was mixed with MMP buffer alone. Prepared solutions were incubated for 24 h at 37°C. After 4, 8, 12, and 24 h, the were withdrawn for further measurements. The cleavage reaction was stopped using ethylenediaminetrtraacetic acid to a final concentration of .24 2.2.Ninhydrin ReactionTo confirm the hydrolysis, the colorimetric reaction with ninhydrin color reagent (4% w/v ninhydrin solution in ethylene glycol monoethyl ether, 200 mM citrate buffer with 0.16 w/v stannous chloride, pH 5.0 at 25°C) was performed. It was then withdrawn and of the samples were mixed with of ninhydrin color reagent and then placed in a boiling water bath for 30 min. Samples were removed and cooled to room temperature. The of isopropanol solution (50% v/v isopropanol in deionized water) was added. Obtained solutions were diluted to 1.5 mL with deionized water and placed in a quartz cuvette and their electronic absorption spectra were measured using Spectrophotometer U-3900, Hitachi, Japan. 2.3.Time-Resolved Fluorescence SpectroscopyIn order to measure the FLT of samples, the time-resolved spectrometer Life Spec II (Edinburgh Instruments Ltd.) with the sub-nanosecond pulsed EPLED diode emitting a light of the wavelength was used. The pulse repetition frequency was 5 MHz. The average output power of the source did not exceed 0.3 to . The spectrometer was equipped with an electronically cooled photomultiplier Hamamatsu R928 connected with a TCC900 PC Card, which incorporates all the electronic modules required for TCSPC. The measurements were carried out with the use of quartz cuvettes at room temperature. Each sample contained of collagen hydrolysis products or nonhydrolyzed collagen with the addition of up to of plasma. The plasma used in the study was obtained from young and healthy subjects without any previous cardiovascular events. The plasma was donated after the subjects received thorough information about the study and gave written consent. The exposure time of the samples was 600 s, when the amount of plasma was , whereas the samples with a higher amount of plasma were measured at 300 s. Such long exposures were necessary to obtain relatively smooth fluorescence decay curves in order to properly analysis of the results. The extended duration of measurement did not affect either the emission spectrum or the fluorescence decay rate. The analysis based on the single-exponential model of decay curves (the least accurate but also the least sensitive to disturbances in the input data) after 30, 90, and 300 or 600 s of exposure gave almost identical results. The optimized values of FLT (obtained for a monoexponential fitting function) did not vary by more than 2% to 3%. The main source of the variation was a different signal/noise ratio present in the fitted data. It suggests that no significant photobleaching/photodegradation process occurred, mainly due to relatively low power and the long wavelength of the excitation beam. It should be mentioned that light of 360 nm was unable to excite fluorescence of tryptophan, tyrosine, and phenylalanine. The MATLAB was used for data analysis and visualization. The FLTs were obtained by deconvolution analysis of the data using the multiexponential model of fluorescence decay and the instrument response function was taken into account. Parameters of the model of fluorescence decay were optimized using the Levenberg–Marquardt algorithm with bound constraints. In order to overcome the local minima problem, the procedure was repeated 200 times with randomly initialized values of the parameters assuming an exponential distribution of lifetimes (with a mean value taken from a single-exponential model) and normalizing the sum of initial fractional intensities of the components. Finally, the best solution in the chi-squared sense was chosen for further analysis. 3.Results3.1.Hydrolyzed Collagen IIITo confirm the hydrolysis, the colorimetric reaction with ninhydrin color reagent was performed and the results were shown in Table 1. According to the ninhydrin reaction, after 7, 11, and 23 h from the beginning of the hydrolysis, similar values of absorbance were received (). Apparently, in less than 11 h, the activity of the enzyme significantly decreased and it is possible that the enzymatic reaction had finished. Table 1Results from ninhydrin color reagent reactions.
The fluorescence decay curves of the samples of hydrolyzed collagen III significantly deviated from a single-exponential function; therefore, in order to obtain the correct fluorescence decay characterization, it was necessary to use the multiexponential fluorescence decay model. The model of three components was used during the analysis of hydrolyzed collagen without the addition of plasma (Fig. 1). However, this model is not efficient if more plasma is used. The real distribution of FLTs in such a system is probably much more complex, especially when one realizes that different conformations of the same specimen may exhibit different deactivations of the excited state. In this work, the authors analyzed only a mean FLT (mFLT) weighted by the fractional contribution of each component to the steady-state intensity calculated from four-exponential models. Fig. 1The decomposition of fluorescence lifetime (FLT) decay on particular components: (a) the signal intensity of hydrolyzed collagen III, (b) the model of three exponential fluorescence decay model, (c) the residuals of the model.  The averaged values of hydrolyzed collagen FLTs after the addition of different amounts of plasma were presented in Fig. 2. When the concentration of plasma was substantial, mFLT increased. Furthermore, when the proportion of hydrolyzed collagen and plasma was , the value of mFLT was highly approximate to the dilution of plasma with physiological saline. The influence of the amount of added plasma was significantly different between hydrolyzed collagen after 4 and 8 h from the beginning of the reaction and after 12 and 24 h. The tendency of the curve was still the same, but the increase of mFLT after a further addition of plasma was relatively slower. The comparison of different amounts of collagen (with respect to the plasma concentration) in samples allowed us to assess the limit of traceability using TRFS in order to detect CDPs. Thus, it is possible to detect CDPs when the CDP concentration in plasma is higher than . When the amount of hydrolyzed collagen in the sample was less than , the increase of mFLT was not observed. CDPs in the presence of the highest amount of plasma did not dominate the signal originating from the fluorescence emission of other proteins. Fig. 2The averaged values of hydrolyzed collagen FLT after addition of plasma; collagen = purified type III collagen from human placenta (Sigma-Aldrich), plasma = human plasma from healthy subject diluted in NaCl: (a) mFLT of samples after 4 h of hydrolysis, (b) mFLT of samples after 8 h of hydrolysis, (c) mFLT of samples after 12 h of hydrolysis, (d) mFLT of samples after 24 h of hydrolysis.  It is necessary to point out that the function of the curves changes in a particular way. At the first time point (), it is the concave-convex function from below, whereas in the next two time points ( and ), the sigmoidal function was observed. In the last time point (), it is the convex function from the bottom. The obtained results look like a function of a dose-response relationship. The changes appear to be regular and depend on the hydrolysis time. The basis for further analysis of the results was the mFLT of the solutions of different proportions of plasma with hydrolyzed collagen as a function of the duration of the hydrolysis (Fig. 3) Additionally, the mFLT of pure hydrolyzed collagen in each time point of the performed hydrolysis was presented. The pure hydrolyzed collagen showed the monotonic increase of the value of mFLT in further steps of hydrolysis. The addition of a small amount of plasma led to an increase of the mFLT of samples obtained at 4 and 8 h of hydrolysis. In further steps of this process, mFLT was substantially shorter, whereas the addition of plasma in the amount of and more had decreased the correlation between the solutions and the time points of the hydrolysis. Fig. 3The mean FLT of the solutions with different proportions of plasma with hydrolyzed collagen as a function of the duration of the hydrolysis; collagen = purified type III collagen from human placenta (Sigma-Aldrich), plasma = human plasma from healthy subject diluted in NaCl: (a) mFLT of samples without addition of plasma, (b) mFLT of samples with 2,5 μl addition of plasma, (c) mFLT of samples with 5 μl addition of plasma, (d) mFLT of samples with 10 μl addition of plasma, (e) mFLT of samples with 20 μl addition of plasma, (f) mFLT of samples with 50 μl addition of plasma, (g) mFLT of samples with 100 μl addition of plasma, (h) mFLT of samples with 600 μl addition of plasma 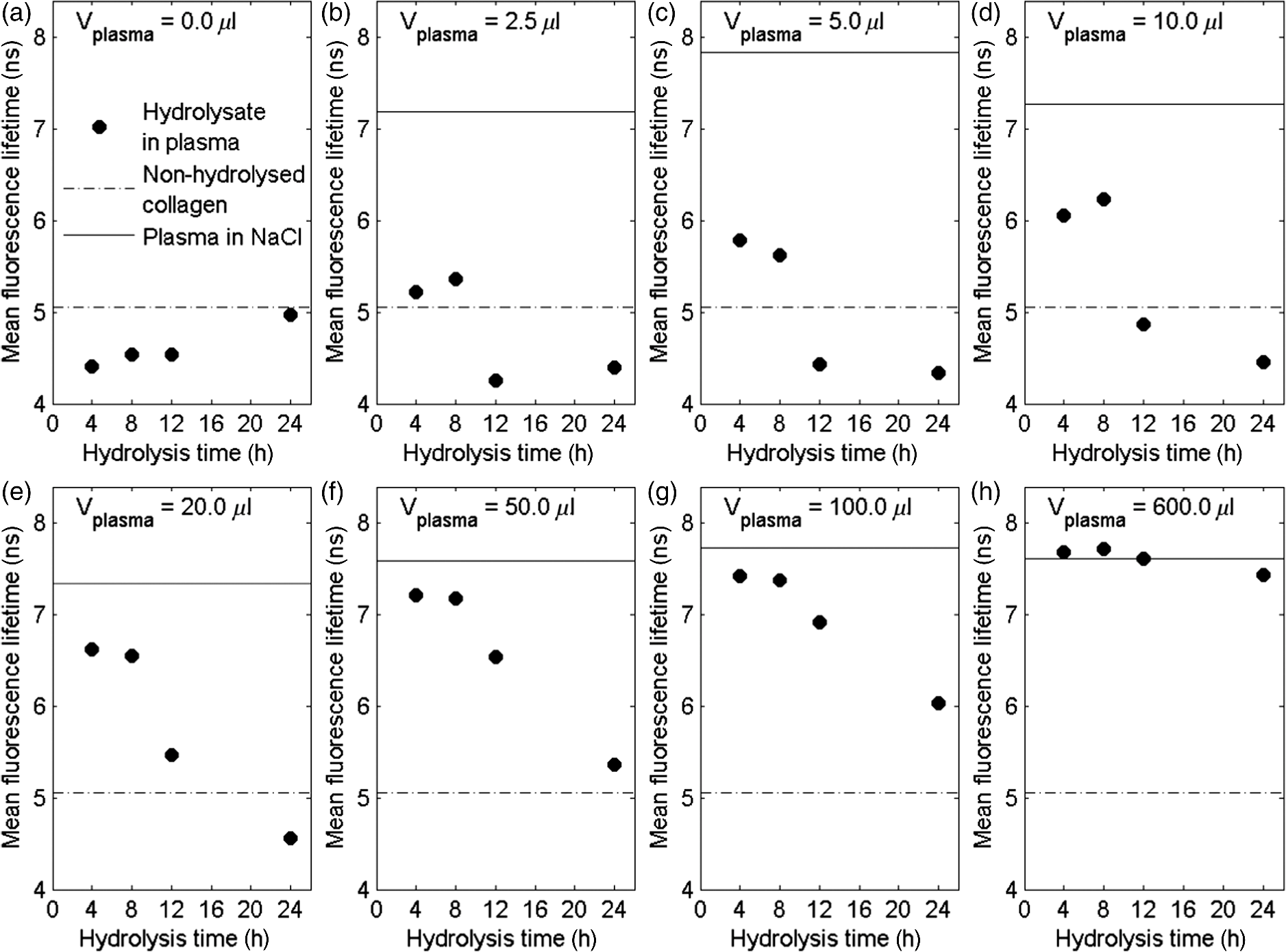 3.2.Study LimitationsThere are a few limitations to the research. To begin, our model of CDPs does not entirely reflect in vivo conditions. With respect to physiology, the activity of CDPs is properly regulated by many factors, especially by the activators and inhibitors, which maintain an appropriate balance between themselves. A potential challenge in future studies is the diversity of proteins in plasma which enhance or attenuate the signal from CDPs. It is important to apply the preliminary fractionation to remove the majority of unnecessary particles from plasma just before the final measurement. In our earlier experiment, we confirmed that the dilution of plasma is not sufficient in this case. It would be problematic to confirm the decomposition of collagen if only the analysis of FLT was used. Additionally, difficulties related to the presence of other proteins, which may affect the FLT, suggest the use of specific spectral probes. The abovementioned spectral probes are designed to be sensitive to the specific interactions with collagen (or CDPs) or to the viscosity of the environment that may be helpful in gaining a deeper insight into the problem. Further studies should still be undertaken. Indeed, it is necessary to explain the mechanism of hydrolysis with the use of two-dimensional electrophoresis and to predict the exact fragments after different steps from the beginning of the hydrolysis. It is also necessary to estimate the accurate molecular weight of CDPs. What should be considered for further research is collagen type I and its degradation products as well as healthy cardiac tissue and cardiac tissue after AMI. These should be studied by creating the subsequent in vitro and in vivo models. The considerable barrier is a technical limitation of the traceability of CDPs. According to our experiment, it is possible to detect them when their concentration in plasma is higher than . 4.DiscussionWe presented data of analysis of FLT due to the complexity of the blood plasma. The biological material contains multiple kinds of proteins and other components concerning the fluorescent properties, which may affect the mFLT. It is extremely difficult to characterize the sample because of the large proportion of albumin, the extensive range in abundance of other proteins as well as the heterogeneity of glycoproteins.25 The endogenous fluorophores which are often used for biological material characterization are aromatic amino acids (tryptophan, tyrosine, phenylalanine), structural proteins (collagen cross-links, elastin), enzyme metabolic co-factors (nicotinamide adenine phosphate dinucleotide and flavin adenine dinucleotide), porphyrins, and lipids.26 Thus, given the multiplicity of the factors determining the fluorescence, analysis of each component could not give reliable information regarding FLT. Our in vitro model of CDPs only partly reflects the in vivo conditions. In the human body, the activity of the used enzyme is strictly regulated by many factors which maintain a balance between ECM deposition and degradation during different physiological processes.27 The activity of MMPs is regulated by transcription, activation, and inhibition.28 Transcription is stimulated by several factors including interleukin 1, platelet-derived growth factor, and tumor necrosis factor . However, it is inhibited by transforming growth factor , heparin, retinoids, and corticosteroids. The activation of pro-MMPs to the MMPs is greatly stimulated by the urokinase plasminogen activator, which is expressed in monocytes and macrophages and is inhibited by tissue inhibitors of metalloproteinase (TIMPs). This system, with a specific receptor and inhibitors plasminogen activator inhibitor (PAI) 1 and 2, regulates the proteolytic activity. The inhibition of activated MMPs by TIMPs as well as drugs, such as tetracyclines and anthracyclines, regulates the proteolysis of ECM.29 The myocardial MMPs and TIMPs are also secreted by several cell types including fibroblasts, inflammatory cells, and endothelial cells. The gene expression is firmly controlled at the transcription level. Several cytokines, polypeptide growth factors, hormones, steroids, and phorbol esters modulate the synthesis and secretion of pro-MMPs and TIMPs.30 Liu et al.31 demonstrated that the collagenases are responsible only for the first degradation step of collagen, in which the fibers are cleaved into specific and fragments. However, gelatinases and cysteine proteases can further degrade the collagen fragments. The data confirmed our results from the ninhydrin reaction because the decreased activity of the enzyme and completion of the reaction was observed earlier than we expected. Kerkvliet et al.32 suggested that MMP1 is responsible for the first degradation step of collagen, and then MMP2 further digests the degraded collagen fibers, whereas Creemers et al.33 demonstrated that MMP2 is responsible for collagen digestion in the soft connective tissues. The precise molecular mechanism of collagen degradation is still inexplicable; however, there are hypotheses which explain the structural changes during this process. Collagen degradation begins with the cleavage of collagen by MMPs at specific sites, which is a covalent bond between glycine and leucine or isoleucine residues, followed by alanine or leucine.34 Therefore, from the analysis of the sequence of target proteins, which fragments will result from the hydrolysis with any of common proteases could be predicted. Due to the collagen triple helical structure, all side chains in collagen are exposed to solvent which differentiates them from globular proteins. Nevertheless, it shows that the covalent bond in the canonical triple helical structure is hidden from the solvent; thus, the collagen triple helix does not fit into the active site of collagenases. This confirms that the existence of locally unfolded states could probably play a major role in the covalent bonding to become available to collagenases.35 Nonetheless, it is essential to explain the specific mechanism of the MMP1 cleavage to conduct further research. The obtained results showed that the increase of added plasma to hydrolyzed collagen extends the mFLT. The visible fluorescence signal from hydrolyzed collagen was observed when a small amount of added plasma was used. This suggests that it is highly likely to detect CDPs in plasma if the interfering proteins were eliminated. According to our experiment, it is possible to detect CDPs when the concentration in plasma is higher than . There are a number of scientific papers,36–39 which confirm the apparition of CDPs in plasma or serum during different disorders. Langberg et al.36 reported that the concentration in plasma of type I collagen degradation product [cross-linked carboxyterminal telopeptide of type I collagen (ICTP)], which is the most common detectable CDPs from type I collagen, after intensive physical training was about . Nurmenniemi et al.37 determined that in patients with IV stage head and neck squamous cell carcinomas, the amount of ICTP was . Rosenquist et al.38 measured the C-terminal telopeptides of type I collagen in serum and detected a concentration of about for premenopausal women and about for healthy postmenopausal women. Perkins et al.39 used Western blotting to check the concentration of circulating precursor forms of type XVIII collagen in acute lung injury patients. The densitometric analysis of gel electrophoresis confirmed increased plasma precursors, especially with the weights of 130 and 170 kDa. Alterations in collagen deposition during scar development in the early phase of wound healing following AMI appear to be associated with the development of infarct expansion and the pathogenesis of myocardial rupture.19 Kasama et al.40 revealed significant correlations between the degree of change in the concentration of PIIINP and echocardiographic exponents of LVR in patients with ST-segment elevation myocardial infarction. Markers of collagen turnover also identify patients with diastolic dysfunction.41,42 Weber et al.43 demonstrated that procollagen peptides as well as MMPs could be used for monitoring cardiac collagen turnover. Iraqi et al.44 showed long-term kinetics of collagen biomarkers in patients with AMI with left ventricle dysfunction and congestive heart failure (HF). Jensen et al.45 confirmed that serum PIIINP remained elevated for more than 4 months after AMI, suggesting ongoing repair processes. Moreover, markers of collagen metabolism (ICTP and PIIINP) reflected reverse left remodeling following cardiac resynchronization therapy in patients with HF.46 These findings suggest that the collagen assessment may assist in identifying patients while they are developing heart remodeling that is still present in a diagnostically challenging condition. For better understanding, the microscopic level of the LVR process in vitro and in vivo models with cardiac tissue should be analyzed. Jugdutt47 studied a canine model of the LVR after AMI. Different models for studying LVR and heart failure development were examined involving large animals (dog, pig, and sheep) and small animals (rabbit, rat, and mouse).48,49 In most models, both types of mRNA (from types I and III collagens) increase in 3 days50 and fall by 7 to 14 days.51 5.ConclusionThe detection method of CDPs based on TRFS may have a significant diagnostic value. However, it is crucial to continue further investigation in this field. Differences between mean FLT from consecutive steps of hydrolysis suggest that it would be possible to determine the degree of collagen degradation. It is essential to further this field of research with the use of cardiac tissue from in vitro and in vivo models, as well as tissue from the human heart. AcknowledgmentsThis study was supported by a research grant from the National Science Centre, Poland (grant No. N N402 497240). ReferencesG. M. Hutchins and B. H. Bulkley,
“Infarct expansion versus extension: two different compilations of acute myocardial infarction,”
Am. J. Cardiol., 41
(7), 1127
–1132
(1978). http://dx.doi.org/10.1016/0002-9149(78)90869-X AJNCE4 0258-4425 Google Scholar
H. F. Weisman et al.,
“Cellular mechanisms of myocardial infarct expansion,”
Circulation, 78
(1), 186
–201
(1988). http://dx.doi.org/10.1161/01.CIR.78.1.186 CIRCAZ 0009-7322 Google Scholar
M. A. Pfeffer and E. Braunwald,
“Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications,”
Circulation, 81
(4), 1161
–1172
(1990). http://dx.doi.org/10.1161/01.CIR.81.4.1161 CIRCAZ 0009-7322 Google Scholar
M. G. Sutton and N. Sharpe,
“Left ventricular remodeling after myocardial infarction: pathophysiology and therapy,”
Circulation, 101
(25), 2981
–2988
(2000). http://dx.doi.org/10.1161/01.CIR.101.25.2981 CIRCAZ 0009-7322 Google Scholar
B. I. Jugdutt,
“Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways,”
Curr. Drug Targets Cardiovasc. Haematol. Disord., 3
(1), 1
–30
(2003). http://dx.doi.org/10.2174/1568006033337276 2212-4063 Google Scholar
G. Olivetti et al.,
“Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats,”
Circ. Res., 67
(1), 23
–34
(1990). http://dx.doi.org/10.1161/01.RES.67.1.23 CIRUAL 0009-7330 Google Scholar
J. B. Caufield and T. K. Borg,
“The collagen network of the heart,”
Lab. Invest., 40
(3), 364
–372
(1979). LAINAW 0023-6837 Google Scholar
B. I. Jugdutt,
“Prevention of ventricular remodeling post myocardial infarction. Timing and duration of therapy,”
Can. J. Cardiol., 9
(1), 103
–114
(1993). CJCAEX 0828-282X Google Scholar
P. Whittaker, D. R. Boughner and R. A. Kloner,
“Role of collagen in acute myocardial infract expansion,”
Circulation, 84
(5), 2123
–2134
(1991). http://dx.doi.org/10.1161/01.CIR.84.5.2123 CIRCAZ 0009-7322 Google Scholar
C. A. Sounders, S. L. K. Bowers and T. A. Baudino,
“Cardiac fibroblast: the renaissance cell,”
Circ. Res., 105
(12), 1164
–1176
(2009). http://dx.doi.org/10.1161/CIRCRESAHA.109.209809 CIRUAL 0009-7330 Google Scholar
M. S. Sacks,
“Incorporation of experimentally-derived fiber orientation into a structural constitutive model for planar collagenous tissue,”
J. Biomech. Eng., 125
(2), 280
–287
(2003). http://dx.doi.org/10.1115/1.1544508 JBENDY 0148-0731 Google Scholar
F. G. Spinale,
“Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function,”
Physiol. Rev., 87
(4), 1285
–1342
(2007). http://dx.doi.org/10.1152/physrev.00012.2007 PHREA7 0031-9333 Google Scholar
R. Visse and H. Nagase,
“Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, biochemistry,”
Circ. Res., 92
(8), 827
–839
(2003). http://dx.doi.org/10.1161/01.RES.0000070112.80711.3D CIRUAL 0009-7330 Google Scholar
G. I. Goldberg et al.,
“Human fibroblast collagenase: complete primary structure and homology to an oncogene transformation-induced rat protein,”
J. Biol. Chem., 261
(14), 6600
–6605
(1986). JBCHA3 0021-9258 Google Scholar
S. Sekita et al.,
“Studies on collagen in the experimental myocardial infarction,”
Jpn. Circ. J., 49 171
–178
(1985). http://dx.doi.org/10.1253/jcj.49.171 JCIRA2 0047-1828 Google Scholar
K. Gelsea, E. Poschlb and T. Aigner,
“Collagens—structure, function, and biosynthesis,”
Adv. Drug. Deliv. Rev., 55
(12), 1531
–1546
(2003). http://dx.doi.org/10.1016/j.addr.2003.08.002 ADDREP 0169-409X Google Scholar
S. Ricard-Blum and F. Ruggiero,
“The collagen superfamily: from the extracellular matrix to the cell membrane,”
Pathol. Biol., 53 430
–442
(2005). http://dx.doi.org/10.1016/j.patbio.2004.12.024 PTBIAN 0369-8114 Google Scholar
K. von der Mark,
“Localization of collagen types in tissues,”
Int. Rev. Connect. Tissue Res., 9 265
–324
(1981). http://dx.doi.org/10.1016/B978-0-12-363709-3.50012-7 ICOTAR 0074-767X Google Scholar
C. Manhenke et al.,
“The relationship between markers of extracellular cardiac matrix turnover: infarct healing and left ventricular remodelling following primary PCI in patients with first-time STEMI,”
Eur. Heart. J., 35
(6), 395
–402
(2014). http://dx.doi.org/10.1093/eurheartj/eht482 EHJODF 0195-668X Google Scholar
M. K. Bode et al.,
“Complete processing of type III collagen in atherosclerotic plaques,”
Arterioscler. Thromb. Vasc. Biol., 19
(6), 1506
–1511
(1999). http://dx.doi.org/10.1161/01.ATV.19.6.1506 ATVBFA 1079-5642 Google Scholar
J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Baltimore, USA
(2006). Google Scholar
J. Kalisz,
“Review of methods for time interval measurements with picoseconds resolution,”
Metrologia, 41
(1), 17
–32
(2004). http://dx.doi.org/10.1088/0026-1394/41/1/004 MTRGAU 0026-1394 Google Scholar
D. P. Millar,
“Time-resolved fluorescence spectroscopy,”
Curr. Opin. Struct. Biol., 6
(5), 637
–642
(1996). http://dx.doi.org/10.1016/S0959-440X(96)80030-3 COSBEF 0959-440X Google Scholar
S. S. Veidal et al.,
“MMP mediated degradation of type VI collagen is highly associated with liver fibrosis—identification and validation of a novel biochemical marker assay,”
PLoS One, 6
(9), e24753
(2011). http://dx.doi.org/10.1371/journal.pone.0024753 1932-6203 Google Scholar
R. Richards-Kortum and E. Sevick-Muraca,
“Quantitative optical spectroscopy for tissue diagnosis,”
Annu. Rev. Phys. Chem., 47 55
–606
(1996). http://dx.doi.org/10.1146/annurev.physchem.47.1.555 ARPLAP 0066-426X Google Scholar
N. L. Anderson and N. G. Anderson,
“The human plasma proteome, history, character, and diagnostic prospects,”
Mol. Cell. Proteomics, 1
(11), 845
–867
(2002). http://dx.doi.org/10.1074/mcp.R200007-MCP200 ATVBFA 1535-9476 Google Scholar
D. E. Gomez,
“Tissue inhibitors of metalloproteinases: structure, regulation and biological function,”
Eur. J. Cell. Biol., 74
(2), 111
–122
(1997). EJCBDN 0171-9335 Google Scholar
D. L. Mann and F. G. Spinale,
“Activation of matrix metalloproteinases in the failing human heart: breaking the tie that binds,”
Circulation, 98
(17), 1699
–1702
(1998). http://dx.doi.org/10.1161/01.CIR.98.17.1699 CIRCAZ 0009-7322 Google Scholar
B. I. Jugdutt,
“Remodeling after infarction and the extracellular collagen matrix: when is enough enough?,”
Circulation, 108
(11), 1395
–1403
(2003). http://dx.doi.org/10.1161/01.CIR.0000085658.98621.49 CIRCAZ 0009-7322 Google Scholar
S. C. Tyagi et al.,
“Co-expression of tissue inhibitor and matrix metalloproteinase in myocardium,”
J. Mol. Cell. Cardiol., 27
(10), 2177
–2189
(1995). http://dx.doi.org/10.1016/S0022-2828(95)91443-9 JMCDAY 0022-2828 Google Scholar
X. Liu et al.,
“A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling,”
J. Cell. Biol., 130
(1), 227
–237
(1995). http://dx.doi.org/10.1083/jcb.130.1.227 JCLBA3 0021-9525 Google Scholar
E. H. M. Kerkvliet et al.,
“Collagen type I, III, and V differently modulate synthesis and activation of matrix metalloproteinases by cultured rabbit periosteal fibroblasts,”
Matrix Biol., 22
(3), 217
–277
(2003). http://dx.doi.org/10.1016/S0945-053X(03)00035-0 MTBOEC 0945-053X Google Scholar
L. B. Creemers et al.,
“Gelatinase a (MMP-2) and cysteine proteinases are essential for the degradation of collagen in soft connective tissue,”
Matrix Biol., 17
(1), 35
–46
(1998). http://dx.doi.org/10.1016/S0945-053X(98)90123-8 MTBOEC 0945-053X Google Scholar
S. Netzel-Arnett et al.,
“Sequence specificities of human fibroblast and neutrophil collagenases,”
J. Biol. Chem., 266
(11), 6747
–6755
(1991). JBCHA3 0021-9258 Google Scholar
C. M. Stultz,
“Localized unfolding of collagen explains collagenase cleavage near imino-poor sites,”
J. Mol. Biol., 319
(5), 997
–1003
(2002). http://dx.doi.org/10.1016/S0022-2836(02)00421-7 JMOBAK 0022-2836 Google Scholar
H. Langberg, L. Rosendal and M. Kjaer,
“Training-induced changes in peritendinous type I collage turnover determined by microdialysis in humans,”
J. Physiol., 534
(1), 297
–302
(2001). http://dx.doi.org/10.1111/tjp.2001.534.issue-1 JPHYA7 0022-3751 Google Scholar
S. Nurmenniemi et al.,
“Type I and III collagen degradation products in serum predict patient survival in head and neck squamous cell carcinoma,”
Oral. Oncol., 48
(2), 136
–140
(2012). http://dx.doi.org/10.1016/j.oraloncology.2011.09.002 EJCCER 1368-8375 Google Scholar
C. Rosenquist et al.,
“Serum CrossLaps One Step ELISA. First application of monoclonal antibodies for measurement in serum of bone-related degradation products from C-terminal telopeptides of type I collagen,”
Clin. Chem., 44
(11), 2281
–2289
(1998). CLCHAU 0009-9147 Google Scholar
G. D. Perkins et al.,
“Type XVIII collagen degradation products in acute lung injury,”
Crit. Care, 13
(2), R52
(2009). http://dx.doi.org/10.1186/cc7779 1364-8535 Google Scholar
S. Kasama et al.,
“Effects of spironolactone on cardiac sympathetic nerve activity and left ventricular remodelling after reperfusion therapy in patients with first ST-segment elevation myocardial infarction,”
Heart, 97
(10), 817
–822
(2011). http://dx.doi.org/10.1136/hrt.2010.215459 HEARFR 1355-6037 Google Scholar
R. Martos et al.,
“Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover,”
Eur. J. Heart Failure, 11
(2), 191
–197
(2009). http://dx.doi.org/10.1093/eurjhf/hfn036 EJHFFS 1388-9842 Google Scholar
R. Martos et al.,
“Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction,”
Circulation, 115
(7), 888
–895
(2007). http://dx.doi.org/10.1161/CIRCULATIONAHA.106.638569 CIRCAZ 0009-7322 Google Scholar
K. T. Weber,
“Monitoring tissue repair and fibrosis from a distance,”
Circulation, 96
(8), 2488
–2492
(1997). CIRCAZ 0009-7322 Google Scholar
W. Iraqi et al.,
“Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure insights from the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (ephesus) study,”
Circulation, 119
(18), 2471
–2479
(2009). http://dx.doi.org/10.1161/CIRCULATIONAHA.108.809194 CIRCAZ 0009-7322 Google Scholar
L. T. Jensen et al.,
“Serum aminoterminal type III procollagen peptide reflects repair after acute myocardial infarction,”
Circulation, 81 52
–57
(1990). http://dx.doi.org/10.1161/01.CIR.81.1.52 CIRCAZ 0009-7322 Google Scholar
S. Umar et al.,
“Myocardial collagen metabolism in failing hearts before and during cardiac resynchronization therapy,”
Eur. J. Heart Failure, 10
(9), 878
–883
(2008). http://dx.doi.org/10.1016/j.ejheart.2008.06.019 EJHFFS 1388-9842 Google Scholar
B. I. Jugdutt,
“The dog model of left ventricular remodelling after myocardial infarction,”
J. Card. Failure, 8
(6), 472
–475
(2002). http://dx.doi.org/10.1054/jcaf.2002.129274 JCFAF9 1071-9164 Google Scholar
B. I. Jugdutt, M. J. Joljart and M. I. Khan,
“Rate of collagen deposition during healing after myocardial infarction in the rat and dog models: mechanistic insights into ventricular remodeling,”
Circulation, 94 94
–101
(1996). http://dx.doi.org/10.1161/01.CIR.94.1.94 CIRCAZ 0009-7322 Google Scholar
K. A. Reimer,
“Animal models for protecting ischemic myocardium: results of the NHLBI Cooperative Study. Comparison of conscious and unconscious dog models,”
Circ. Res., 56 651
–665
(1985). http://dx.doi.org/10.1161/01.RES.56.5.651 CIRUAL 0009-7330 Google Scholar
D. Chapman, K. T. Weber and M. Eghbali,
“Regulation of fibrillary collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium,”
Circ. Res., 67
(4), 787
–794
(1990). http://dx.doi.org/10.1161/01.RES.67.4.787 CIRUAL 0009-7330 Google Scholar
F. J. Villarreal and W. H. Dillman,
“Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta I, fibronectin and collagen,”
Am. J. Physiol., 262
(6), H1861
–H1866
(1992). AJPHAP 0002-9513 Google Scholar
BiographyJoanna Sikora obtained a master’s degree (2013) in biotechnology with a specialty of medicine at Nicolaus Copernicus University, Collegium Medicum, in Bydgoszcz. She is currently a PhD student in medical sciences at Nicolaus Copernicus University, Collegium Medicum in Bydgoszcz, and an assistant in the Department of Biochemistry, Faculty of Medicine, Nicolaus Copernicus University, Collegium Medicum in Bydgoszcz. Her scientific interests concern fluorescence spectroscopy, acute coronary syndromes, and analysis of proteins by two-dimensional electrophoresis. Michał Cyrankiewicz obtained a master’s degree (2003) in technical physics at the University of Science and Technology in Bydgoszcz and received his PhD diploma (2011) from Poznan University of Technology. He is currently an assistant professor at the Biophysics Department’s Faculty of Pharmacy at Nicolaus Copernicus University, Collegium Medicum in Bydgoszcz. His scientific interests concern optical properties of metallic nanoparticles and their applications in surface enhanced Raman and fluorescence spectroscopy. Tomasz Wybranowski completed studies in the field of technical physics with a specialty in computerized physical measurement from the Faculty of Chemical Technology and Engineering at the University of Science and Technology in Bydgoszcz. In 2006, he was granted the degree of master of science, engineer. In 2014, he obtained the degree of PhD in medical sciences from the Faculty of Medicine, Nicolaus Copernicus University, Collegium Medicum, in Bydgoszcz. His main scientific interests include optical spectroscopy, nanoparticles, and oxidative stress. Blanka Ziomkowska received a master’s degree in physics from the Faculty of Physics, Astronomy, and Informatics of Nicolaus Copernicus University in 2001. In 2009, she obtained a PhD in medical sciences from the Faculty of Medicine at Nicolaus Copernicus University in Bydgoszcz. Her main scientific interests are fluorescence spectroscopy methods and the interaction of drugs with blood components. Borys Ośmiałowski obtained his diploma (1997) in chemistry at the Department of Chemistry and Chemical Engineering (Bydgoszcz), his PhD diploma (2003) at the Department of Chemistry at Nicolaus Copernicus University (Toruń), and habilitation (2012) at University of Łódź. From 2001 to 2002, he was a holder of the Foundation for Polish Science grant, and in 2004, he had a fellowship for a postdoctoral stay at the University of Zurich (in prof. Jay Siegel’s group). Main scientific interests include supramolecular chemistry, fluorescent probes, and tautomerism. Ewa Obońskas graduated in 2012 from the Faculty of Medicine at Nicholaus Copernicus University, Collegium Medicum, in Bydgoszcz. She is currently a resident in the Department of Cardiology and Internal Medicine, University Hospital No 1 in Bydgoszcz. She works as an assistant in the Pharmacology Department at Nicholaus Copernicus University, Collegium Medicum, in Bydgoszcz. Main scientific interests include interventional cardiology and acute coronary syndromes. Beata Augustyńska received a PhD (2009) in medical sciences from the Faculty of Medicine at Nicholaus Copernicus University, Collegium Medicum, in Bydgoszcz, and habilitation in 2012 at Nicholaus Copernicus University in Toruń. She is currently a head of the Department of Biochemistry, Faculty of Medicine, Nicolaus Copernicus University, Collegium Medicum, in Bydgoszcz. Main scientific interests include oxidative stress, and enzymatic and nonenzymatic markers of oxidative stress in pathological states. Stefan Kruszewski received a PhD (1986) in physics at Nicolaus Copernicus University in Toruń and habilitation in surface physics in 1999 at Wroclaw Technical University. From 1998 to 2001, he worked as a research associate at the College of Pharmacy, University of Kentucky in Lexington (USA). In 2013, he was appointed professor of pharmacy. His areas of expertise included Raman and SERS spectroscopy, and fluorescence spectroscopy methods applied to determine the properties of drugs. Jacek Kubica received a degree in medicine in 1987 and a PhD in medical sciences in 1992 from the Medical Faculty at the Medical University of Gdańsk. In 2013, he was appointed as an associate professor at Nicolaus Copernicus University, Collegium Medicum, in Bydgoszcz. He is currently a head of the Department of Cardiology and Internal Medicine, Faculty of Medicine, Nicolaus Copernicus University, in Bydgoszcz. He is a specialist of interventional cardiology. His areas of expertise include antiplatelet therapy and acute coronary syndromes. |



