1.IntroductionTraumatic brain injury (TBI) remains the most common cause of death in persons under age 45 in the Western world. The societal impact is profound, with 2 million cases, 220,000 hospitalizations, and 52,000 deaths from head trauma occurring each year in the United States.1 2 One of the principal determinants of morbidity and mortality following TBI is traumatic axonal injury (TAI). While multiple tissue and animal studies have proposed various pharmacological or physiological interventions to reduce axonal injury, these studies have not translated into novel therapeutic options in the clinical setting.3 The development of new methods for detecting protein interactions and elucidation of critical mechanisms of injury in animals and humans will greatly enhance the transition of promising treatments from benchtop to bedside. Current hypotheses on the pathogenesis of TAI, based on previous immunohistochemical and histochemical studies, involve activation of apoptotic cascades secondary to TBI (Ref. 4). A pivotal element of these hypotheses involves5 6 a potential interaction between the proapoptotic protein BAD and prosurvival protein Bcl-xL. The interaction between BAD and Bcl-xL provide a theoretical link between studies demonstrating overt axolemmal disruption with presumed loss of ionic homeostasis and studies demonstrating mitochondrial disruption with cytochrome c release and activation of caspase proteolytic enzymes in relation to local axonal breakdown.7 This proposed theoretical link involves the Ca 2+ -mediated activation of second messengers such as calmodulin secondary to the loss of ionic homeostasis across a traumatically perturbed axolemma. Targets of calmodulin include the phosphatase calcineurin, which can dephosphorylate and activate the proapoptotic protein BAD. In in vitro models of apoptosis and models of spinal cord injury, dephosphorylated BAD has been shown to translocate to the mitochondria and bind to the prosurvival protein Bcl-xL, leading to the breakdown of mitochondria.8 As mitochondrial breakdown with release of cytochrome c accompanied by activation of the caspase cascade of proteolytic enzymes has been demonstrated in relation to local axonal breakdown,7 9 calcineurin-mediated dephosphorylation of BAD with displacement of Bcl-xL has been hypothesized to play a role in traumatic axonal pathogenesis. Current techniques for exploring protein interactions in tissue are limited. Techniques such as coimmunoprecipitation, affinity chromatography, and two-hybrid yeast systems require the homogenization of gross samples of tissue. Thus, while providing information on overall changing trends of protein interactions, they provide little information on how protein interactions occur at the individual cellular level. Neither conventional immunohistochemical/histochemical nor ultrastructural approaches have the capacity to shed insight on whether BAD and Bcl-xL interact in TAI in vivo. In this paper, we describe an approach for employing fluorescence resonance energy transfer (FRET) microscopy in tissue in an in vivo model of neurological disease using aldehyde-fixed rat brain tissue sections. FRET is a quantum mechanical property detected by light microscopy involving radiationless energy transfer from an excited donor fluorophore to an acceptor fluorophore directly through resonance interactions across limited distances.10 While FRET microscopy has yielded significant insights in vitro in determining protein interactions in cell culture preparations,11 12 13 it is a tool that remains relatively unexplored in vivo with tissue analysis. Specifically, the current investigation employs confocal and two-photon excitation FRET microscopy to explore protein interactions in TAI following TBI. While there are in vitro models to assess the effect of stress and strain on individual axons and neuronal cell bodies following TBI, the full spectrum of injury resulting from the complex biomechanical interactions among glia, neurons, axons, and the extracellular milieu cannot be adequately assessed in vitro. As such, the capacity to employ FRET microscopy in studying TAI following TBI constitutes a significant advance in determining the pathogenesis of TAI. 2.Materials and Methods2.1.Animal SurgeryAdult male Sprague-Dawley rats were subjected to an impact acceleration injury described previously in detail.14 Specifically, rats weighing between 350 and 400 g received induction anesthesia using 5 Isoflurane in a bell jar for 8 to 10 min. Following anesthesia induction, animals were endotracheally intubated and maintained on 3 Isoflurane in 30 O 2 and 70 N 2 O using a modified medical anesthesia machine. The animals were then shaved and prepared in sterile fashion for surgery, followed by subcutaneous injection of 1 ml of 0.25 bupivicane into the planned incision site. A 3-cm midline incision in the scalp was then made, and periostial membranes were separated, exposing bregma and lambda. A metal disk 10 mm in diameter and 3 mm thick was attached to the skull with cyanoacrylate and centered between bregma and lambda. Isoflurane was stopped until animals regained respiratory and hindleg reflexes. The animal was then placed prone on a foam bed with the metal disk directly under a Plexiglas tube. A 450-g brass weight was dropped through the tube from a height of 2 m, striking the disk. The animal was then ventilated on 100 O 2 while the skull was inspected for fracture, which would eliminate the animal from the study, and the incision was sutured with 3-0 Vicryl sutures. When the animal recovered spontaneous respirations, the endotracheal tube was removed and the animal was returned to its cage for postoperative observation. Sham surgery animals underwent an identical procedure with the exception of the weight striking the metal disk. All procedures involving live animals were approved by the Institutional Animal Care and Use Committee of the University of Virginia, and were performed according to the principles of the Guide for the Care and Use of Laboratory Animals, published by the Institute of Laboratory Resources, National Research Council (NIH publication 85-23—2985). 2.2.Tissue PreparationSix hours following injury, or 1 h following sham injury, animals were euthanized with a lethal dose injection of 0.5-ml Ketamine and 0.5-ml Xylazine. The animals were then immediately perfused transcardially with 200 ml cold 0.9 saline to wash out all blood. This was followed by 4 paraformaldehyde in Millonigs buffer, initially at 100 ml/min for 2 min, then at 20 ml/min for 40 min. The entire brain, brainstem, and rostral spinal cord was removed and immediately placed in 4 paraformaldehyde for 24 h. Following 24 h of fixation, the brain was blocked by cutting the brainstem above the pons, cutting the cerebellar peduncles, and then making sagittal cuts lateral to the pyramids. The resulting tissue containing the corticospinal tracts and the medial lemnisci, areas shown previously to yield traumatically injured axons, was then sagitally cut on a vibratome into sections either 40, 60, 80, or 100 μm thick. These sections were serially placed in phosphate buffered saline (PBS) in 12-well plates. The tissue was washed three times in PBS, followed by 1 h in 1 hydrogen peroxide in PBS to eliminate endogenous peroxidase activity, followed by three additional washes in PBS. The tissue underwent temperature-controlled microwave antigen retrieval with our previously described technique.15 Specifically, the tissue was placed in citric acid buffer and then placed in a laboratory grade microwave with a temperature probe and two beakers of ice water to reduce thermal effects. The tissue was microwaved for 5 min at 70 power, with the probe in an additional well filled with an equal volume of distilled water, such that if the temperature exceeded 45 °C, the microwave power was suspended until the temperature was below 45 °C. The tissue was then removed from the microwave for 5 min, during which time ice packs were placed in the microwave. The tissue then underwent an additional 5 min of microwaving, again using beakers of ice water and the temperature probe. The tissue was then allowed to cool for 20 min on a shaker plate, followed by three rinses in PBS. The tissue was preincubated in a solution containing 10 normal goat serum (NGS) and 0.2 Triton X in PBS for 40 min. 2.3.ImmunohistochemistryThe tissue was incubated in polyclonal antibody raised in rabbit against Bcl-xL amino acids 126 to 188 (sc-7195, Santa Cruz Biotechnology) at a dilution of 1:200 in 1 NGS in PBS overnight at 4 °C. Following incubation in primary antibody, the tissue was washed three times in 1 NGS in PBS, then incubated in a secondary anti-rabbit IgG antibody conjugated with Alexa 555 fluorophore (Molecular Probes) for 2 h. The tissue was washed three times in 1 NGS in PBS, and preincubated in 10 NGS for 40 min. The tissue was then incubated in the second primary antibody, monoclonal anti-BAD amino acids 1 to 168 raised in mouse (sc-8044, Santa Cruz Biotechnology) at a dilution of 1:50 in 1 NGS in PBS overnight at 4 °C. The tissue was again washed three times in the 1 NGS solution, and incubated in a secondary anti-mouse IgG antibody conjugated with Alexa 488 (Molecular Probes) for 2 h. The tissue underwent a final wash in 0.1 M phosphate buffer and then was mounted using an antifade agent (Molecular Probes) and coverslipped. The slides were sealed with acrylic and stored in the dark in a laboratory refrigerator. 2.4.ControlsSeveral dilution studies and immunohistochemical control studies were conducted both prior to and during the experimental protocol. Initially, primary antibodies were used in a brightfield dilution study to determine antibody concentrations with optimum signal intensity with minimal background staining. Tissue was prepared using the described protocol, including microwave antigen retrieval and 1-h wash in 1 H 2 O 2, and incubated in various dilutions of primary antibody overnight. The tissue then underwent secondary labeling with a 1:200 dilution of species-specific biotin labeled secondary antibody (Vector, Burlingame, California), reacted with an avidin-horseradish peroxidase complex (ABC Standard Elite Kit, Vector Labs). The tissue was incubated for 12 min in a solution of 0.05 diaminobenzidine, 0.01 H 2 O 2, and 0.3 imidazole in 0.1 M phosphate buffer to create a visible reaction product. The tissue was mounted on glass slides, coverslipped, and examined using a Nikon Eclipse 800 microscope. Fluorescent-labeled dilution studies were likewise conducted to determine optimal antibody concentrations of monoclonal BAD and polyclonal Bcl-xL. Following antigen retrieval and a 1-h wash in 1 H 2 O 2, the tissue was incubated overnight in monoclonal BAD antibody or polyclonal Bcl-xL at dilutions ranging from 1:50 to 1:2000. Tissue was then washed in 1 NGS and incubated in secondary antibody, specifically anti-rabbit IgG labeled with Alexa 555 for the Bcl-xL and anti-mouse IgG labeled with Alexa 488 for the BAD. The tissue was mounted on glass slides using an antifade agent (Molecular Probes), coverslipped, and sealed with acrylic. The tissue was then examined and images acquired using the laser scanning confocal microscope system described later. The optimal dilution was determined both by selecting for high signal to noise ratio and for approximately equal fluorescent signal intensity at equal laser excitation powers. Immunofluorescent control studies were also undertaken to ensure that cross-reactivity did not exist between the secondary antibodies and the tissue, and to ensure that the species-specific secondary antibodies did not label the other primary antibody epitopes. Following tissue preparation as described, specimens were incubated in either BAD antibody raised in mouse at a dilution of 1:50, Bcl-xL antibody raised in rabbit at a dilution of 1:200, or no primary antibody. The mouse anti-BAD was then incubated in a 1:200 dilution of Alexa 555 anti-rabbit IgG, the rabbit anti-Bcl-xL was incubated in a 1:200 dilution of Alexa 488 anti-mouse IgG, and specimens without primary label were incubated in both secondary antibodies. Slides were prepared and examined using the confocal FRET (C-FRET) microscopy described later. Precision FRET (PFRET) analysis requires the acquisition of images from tissue labeled with either donor fluorophore or acceptor fluorophore alone. Simultaneous with the double labeling of tissue, the single labeled specimens were processed using identical tissue. For the secondary antibody donor labeled specimens, the tissue underwent the described procedure including incubation in BAD monoclonal antibody; however, the anti-mouse IgG antibody labeled with Alexa 488 was eliminated and instead the tissue was incubated in 1 NGS for 2 h. Likewise, for secondary antibody acceptor labeled specimens, the tissue was incubated in both primary antibodies, but the secondary anti-rabbit IgG antibody labeled with Alexa 555 was eliminated. 2.5.C-FRET and Two-Photon FRET Microscopy System ConfigurationThe system consists of a Nikon TE300 epifluorescent microscope with a 100-W Hg arc lamp. A Plan Fluor 20× numerical aperture (NA) 0.75 MIMM objective lens was used for both C-FRET and two-photon FRET (2p-FRET) image acquisition. TE300 was coupled to a Biorad Radiance 2100 confocal/multiphoton system (www.cellscience.bio-rad.com). A 10-W Verdi pumped, tunable (model 900 Mira, www.coherentinc.com) modelocked ultrafast (78 MHz) pulsed (<150 fs) laser was coupled to the laser port of a Radiance 2100. This laser is equipped with x-wave optics for easy tunable range of the entire wavelength (700 to 1000 nm). The system was equipped with a laser spectrum analyzer (Model E201; www.istcorp.com) to monitor the excitation wavelength and a power meter to measure the laser power at the specimen plane (Model SSIM-VIS & IR; www.coherentinc.com). The Radiance system was equipped with an external detector and four internal detectors for fluorescence imaging. The transmission detector was used for transmission imaging and also for second-harmonic generation (SHG) imaging. LaserSharp 2000 software was used to acquire both C-FRET and 2p-FRET images using the internal detectors. For C-FRET, an argon laser emitting at 488 nm was used to excite the donor fluorophore, while a HeNe green laser emitting at 543 nm was used to excite the acceptor fluorophore. Emissions from the various fluorophores were split using a 560-nm dichroic mirror and filtered using an HQ528/30-nm filter for the donor emission channel, and an HQ590/70-nm filter for the acceptor channel (www.chromatech.com). For 2p-FRET microscopy, following confocal laser scanning image acquisition, an inline tunable Ti:sapphire laser was used to excite the donor fluorophore at 790 nm and at 730 nm for the acceptor fluorophore, and emissions were captured using the same filters as for confocal microscopy. A key benefit of 2p-FRET microscopy is the ability to acquire images from tissue specimens greater than 40 μm thick, the normal limits of C-FRET microscopy. Immunohistochemical labeling methods, however, have a disadvantage in labeling thick tissue, as adequate diffusion of antibodies is generally limited to the outer 10 μm of tissue. Therefore, to determine if images acquired using 2p-FRET microscopy in thicker tissue specimens were appropriate for FRET image analysis, we utilized the z-plane focus motor of the microscope to focus on the distal surface of the specimen. 2.6.FRET Data AnalysisOne of the important conditions for FRET to occur is the overlap of the emission spectrum of the donor with the absorption spectrum of the acceptor.16 As a result of spectral overlap, the FRET signal is always contaminated by donor emission into the acceptor channel and by the excitation of acceptor molecules by the donor excitation wavelength. Both of these signals are termed spectral bleedthrough (SBT) signal into the acceptor channel. In principle, the SBT signal is same for C-FRET and 2p-FRET microscopy. In addition to SBT, the FRET signals in the acceptor channel also require correction for spectral sensitivity variations in donor and acceptor channels, autofluorescence, and detector and optical noise, which contaminate the FRET signal. Seven images were acquired using the appropriate filters as described for PFRET analysis.16 The data analysis algorithm works under the assumption that the double-labeled cells and single-labeled donor and acceptor cells, imaged under the same conditions, provide the same SBT dynamics. The algorithm described in the literature follows fluorescence levels pixel by pixel to establish the level of SBT in the single-labeled cells, and then applies these values as a correction factor to the appropriate matching pixels of the double-labeled cell.16 Precision FRET then is where uncorrected FRET (UFRET/f), ASBT is the acceptor SBT signal, and DSBT is the donor SBT signal, established in the corresponding single-labeled cells.The acquired seven images in C-FRET and 2p-FRET microscopy systems were processed using the PFRET 1.0 software (www.circusoft.com) developed based on the algorithm already described and in the literature.16 This software also enables estimating the energy transfer efficiency (En) and the distance (rn) between donor and acceptor molecules using these equations.16 3.ResultsThe development of a tissue FRET methodology required refinement of three processes, including immunofluorescent labeling of tissue with antibodies directed toward donor and acceptor molecules, acquisition of images using C-FRET and 2p-FRET microscopy, and software-aided pixel-by-pixel analysis of the resulting images. As already described in Sec. 2, we performed a series of preliminary immunohistochemical dilution and control experiments to determine the appropriate working antibody concentrations and to verify the absence of nonspecific or cross-reactive interactions that would confound our data analysis. Brightfield dilution studies with BAD and Bcl-xL antibodies revealed ubiquitous labeling, a 1:200 dilution of BAD intensely labeled soma, and moderately labeled axons, while a 1:500 dilution of Bcl-xL intensely labeled both axons and soma. Fluorescent dilution studies revealed similar results, a 1:50 dilution of BAD antibody labeled with a 1:200 dilution of anti-mouse Alexa 488 IgG revealed moderate labeling of axons and soma, a 1:200 dilution of Bcl-xL antibody labeled with a 1:200 dilution of anti-rabbit Alexa 555 IgG intensely labeled both axons and soma (Fig. 1). Figure 1BAD and Bcl-xL heterodimerization. The heterodimerization of BAD labeled with Alexa 488 and Bcl-xL labeled with Alexa 555 approximates the fluorophores within 100 Å of each other, enabling fluorescent resonance energy transfer.  Control fluorescent immunohistochemical studies were performed to ensure that cross-reactivity did not exist between the secondary antibodies and the tissue, and to ensure that the species-specific secondary antibodies did not label the other primary antibody epitopes. Tissue incubated in the secondary anti-rabbit and anti-mouse antibodies at 1:200 dilutions revealed no labeling of axons or soma. Likewise, tissue incubated in either mouse-BAD or rabbit-Bcl-xL, and then labeled with the opposite species secondary antibody revealed no labeling of axons or soma. Following determination of the optimal dilutions, we prepared specimens for PFRET analysis. Using serial sections of tissue, we simultaneously labeled injured tissue with donor only (BAD/Alexa 488), acceptor only (Bcl-xL/Alexa 555), and with both donor and acceptor. The specimens were then examined for axons demonstrating vacuolization or formation of retraction bulbs, morphological characteristics of axonal injury. Using the laser scanning confocal microscope (LSCM) with the excitation lasers and emission filters as described above, the seven images (Fig. 2) (see Color Plate 1) of these axons required for PFRET analysis were collected as shown in Fig. 2. The uncorrected FRET image [Fig. 2(f)] underwent image correction for DSBT and ASBT, resulting in the PFRET image [Fig. 2(h)]. Multiple regions of interest (ROIs) were then drawn within the imaged axon, the efficiency of energy transfer, and the estimated distance between fluorophores was calculated for each ROI with PFRET software, and the mean and standard deviation of these values were calculated. Figure 2Seven images required for PFRET analysis. Six-hour-postinjury tissue was prepared with mouse anti-BAD IgG 1 labeled with Alexa 488 anti-mouse antibody (donor) and/or rabbit anti-Bcl-xL IgG labeled with Alexa 555 anti-rabbit IgG (acceptor): (a) donor labeled, donor excitation, donor channel; (b) donor labeled, donor excitation, acceptor channel; (c) acceptor labeled, donor excitation, acceptor channel; (d) acceptor labeled, acceptor excitation, acceptor channel; (e) double labeled, donor excitation, donor channel; (f) double labeled, donor excitation, acceptor channel; (g) double labeled, acceptor excitation, acceptor channel; and (h) PFRET image following processing as described. Calculated mean energy transfer efficiency was 22.4±2.2 and estimated distance was 83.0±1.7 Å. Histograms represent pixel intensity for identical ROI for uncorrected FRET (i) and PFRET (j) images. Bar=10 μm. Color Plate 1 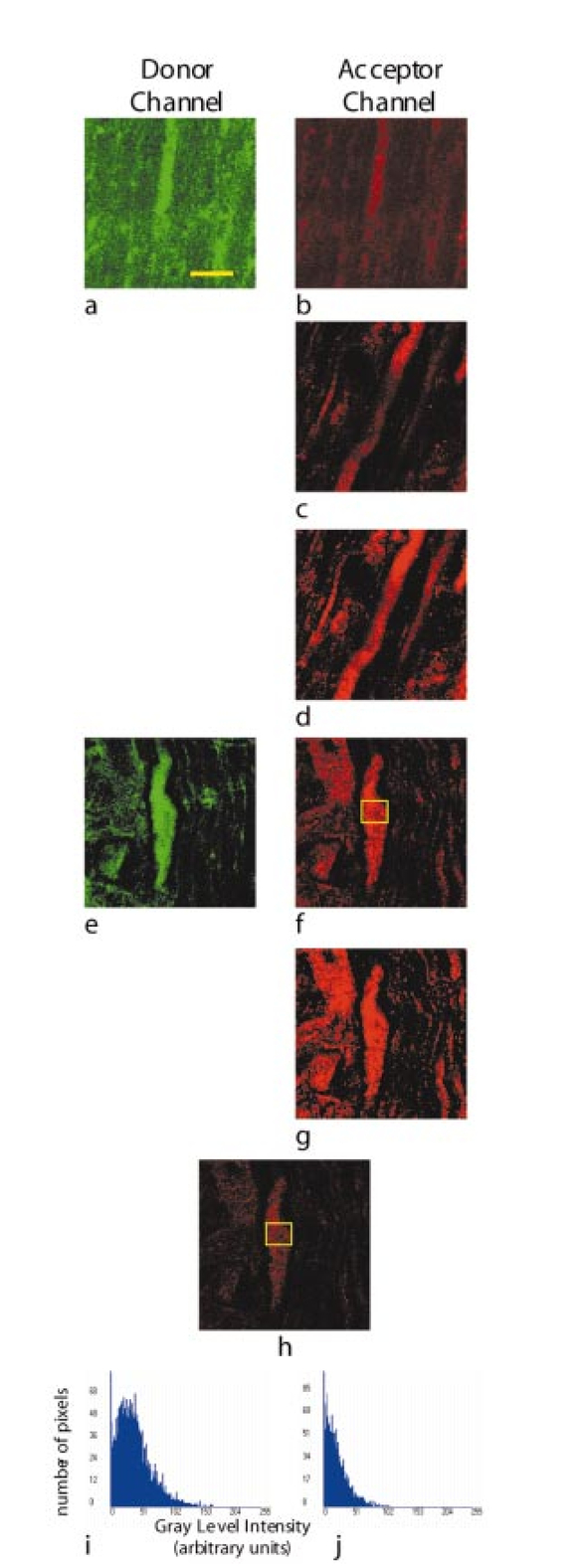 Based on previous experiments, we hypothesized that following traumatic axonal injury, increased levels of intracellular Ca 2+ should result in the activation of calcineurin, subsequent dephosphorylation of BAD, and translocation of BAD to the mitochondria, followed by the proapoptotic binding of BAD to Bcl-xL. According to this hypothesis, prior to injury, BAD and Bcl-xL should remain separate, the distance between them should be greater than 100 Å, and no FRET signal should be detected. Conversely, following injury, if heterodimerization exists between BAD and Bcl-xL, the distance between them should be less than 100 Å, resulting in FRET signal detection. In tissue processed from animals receiving a sham injury, we observed axons with normal morphologies and ubiquitous labeling with both BAD and Bcl-xL. Images for PFRET analysis (Fig. 3) (see Color Plate 1) were acquired using the LSCM system already described above, including images from both the donor [Fig. 3(a)] and acceptor [Fig. 3(b)] channel in the double-labeled tissue excited at 488 nm. The images were then processed using the image analysis software to produce a PFRET image [Fig. 3(c)], ROIs were drawn, and the mean energy transfer efficiency was 7.24±1.9, and an estimated distance greater than 100 Å (Fig. 6). Figure 3Laser scanning confocal images acquired from control (a) to (c) or injured animals at 6 h postinjury (d) to (f). Following immunohistochemical processing with mouse anti-BAD IgG 1 labeled with Alexa 488 anti-mouse antibody (donor) and rabbit anti-Bcl-xL IgG labeled with Alexa 555 anti-rabbit IgG (acceptor), images required for PFRET analysis were acquired. In sham injured animals, axons demonstrate ubiquitous labeling for BAD (a), low uncorrected acceptor emission (b), and low efficiency of transfer following PFRET image correction (c). In axons from animals 6 h postinjury, BAD likewise demonstrates ubiquitous labeling of axons (d), however, significant acceptor emission occurs with donor excitation (e), and PFRET image correction demonstrates significant energy transfer (f) with an efficiency of 23.1±2.1 and estimated distance of 82.5±2.6 Å. Bar=10 μm.  In contrast to our findings in noninjured animals, tissue processed from animals allowed to survive for 6 h after injury demonstrated ubiquitously labeled axons with vacuolization and formation of retraction bulbs. Images of these axons were acquired, including the donor channel [Fig. 3(d)] and uncorrected FRET [Fig. 3(e)] images, and a PFRET image [Fig. 3(f)] was produced. ROIs were drawn, and the mean energy transfer was 29.03±6.63, significantly greater than the noninjured animals (p<0.01), with an estimated distance of approximately 78.7±4.2 Å (Fig. 6). Two-photon microscopy has advantages over confocal microscopy, including reduced focal volume and less light scattering in specimen.17 18 To determine if images acquired using 2p-FRET microscopy would demonstrate FRET efficiencies similar to C-FRET, we utilized the same confocal microscope setup coupled with an inline tunable Ti:sapphire laser. With this setup, we were able to acquire confocal images, then switch to 2p microscopy and acquire images of the same axons in the same focal plane (Fig. 4). Figure 4Multiphoton images acquired from animals subjected to a sham injury (a) to (c) or 6 h postinjury (d) to (f). Following immunohistochemical processing with mouse anti-BAD IgG 1 labeled with Alexa 488 anti-mouse antibody (donor) and rabbit anti-Bcl-xL IgG labeled with Alexa 555 anti-rabbit IgG (acceptor), images required for PFRET analysis were acquired. In sham injured animals, axons demonstrated ubiquitous labeling for BAD with excitation with a multiphoton laser tuned to 790 nm (a), low uncorrected acceptor emission with 790-nm multiphoton excitation (b), and low efficiency of transfer following PFRET image correction (c). In axons from animals 6 h postinjury, BAD likewise demonstrates ubiquitous labeling of axons (d), however, significant acceptor emission occurs with donor excitation (e), and PFRET image correction demonstrates energy transfer from donor to acceptor with calculated efficiency of 32.4±6.5 and estimated distance of 76.5±3.8 Å. Bar=10 μm. 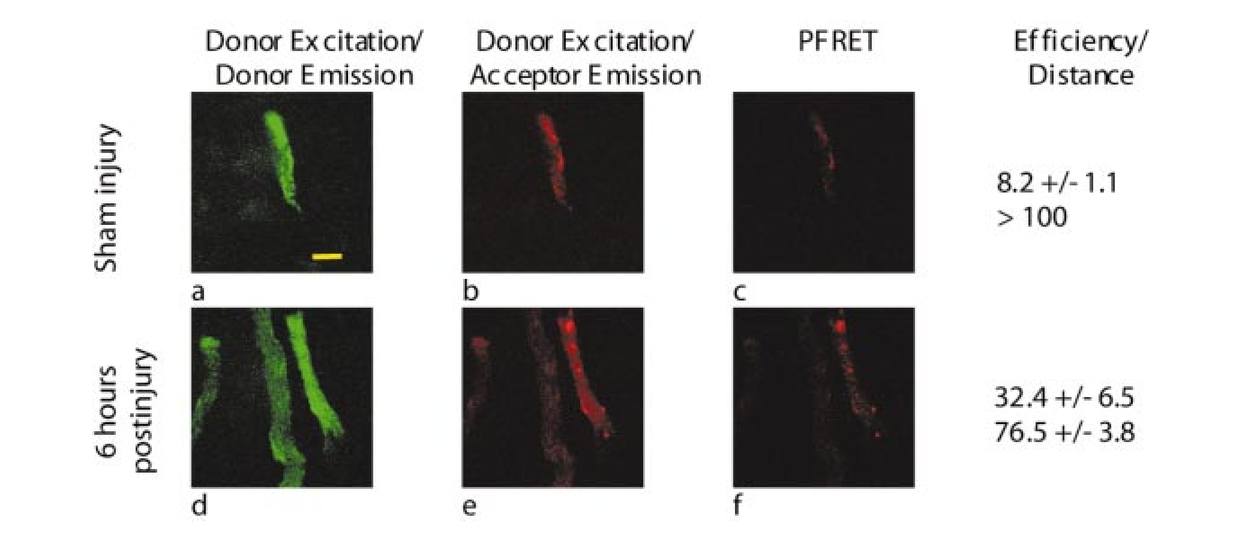 In tissue from animals receiving a sham injury, we observed normal axonal profiles with ubiquitous labeling of BAD and Bcl-xL. Images of these axons were acquired, including donor channel emission image [Fig. 4(a)] and uncorrected FRET image [Fig. 4(b)] in double-labeled tissue excited with a 790-nm laser (see Color Plate 2). Donor and acceptor excitation wavelength was selected according to the methodology described in the literature.16 Images were processed using image analysis, resulting in the PFRET image [Fig. 4(c)]. ROIs were drawn, the mean transfer of energy was calculated at 8.2±1.1 and the estimated distance was greater than 100 Å. In tissue from 6-h postinjury tissue, we observed vacuolated axon segments and sporadic retraction bulbs. Images of these axons were acquired, including donor channel [Fig. 4(d)] and uncorrected FRET [Fig. 4(e)]. and the PFRET image [Fig. 4(f)] was produced. These axons demonstrating morphological characteristics of injury had mean energy transfers greater than 32.4±6.5, and estimated distance of 76.5±3.8 Å (Fig. 6). Figure 6Bar graphs of FRET efficiency between fluorophore labeled BAD and Bcl-xL at 6 h postinjury. FRET efficiencies (a) of both confocal and 2p microscopy were significantly (*,p<0.01) greater at 6 h postinjury compared to sham injured animals. There was no significant difference in efficiencies between confocal or 2p methods in either sham (p=0.34) or at 6 h postinjury (p=0.24) Estimated distance (b) between BAD and Bcl-xL was significantly (*,p<0.01) decreased at 6 h, as compared to sham injured. There was no significant difference between confocal or 2p methods in distance estimations.  An additional advantage of 2p-FRET microscopy is the ability to penetrate tissue specimens, and visualize deep focal planes. To determine if we could image axons in tissue sections greater than 40 μm, we prepared tissue specimens 60, 80, and 100 μm thick with an adjustable vibratome. After immunohistochemical labeling of the specimens, we imaged the proximal surface of the specimens using C-FRET microscopy. Then, using a calibrated z-focus motor, we focused on the distal surface of the specimen by changing the z-plane focus 60, 80, or 100 μm, respectively. Using the tunable Ti:sapphire laser, the images required for FRET analysis were acquired (Fig. 5) (see Color Plate 2). Axons ubiquitously labeled for both BAD and Bcl-xL were visualized in all tissue specimens. However, the relative number of axons seen decreased as the thickness increased. Likewise, the amount of background fluorescence decreased as the thickness increased. This suggests that as the thickness of the tissue increased, the emission signal decreased, and we therefore were able to visualize only the most intensely labeled axons. In 6-h-postinjury tissue, vacuolated axons were imaged for PFRET analysis, ROIs were drawn, mean efficiencies calculated, and the distances estimated. At 60, 80, and 100 μm, we obtained FRET efficiencies greater than 20, and estimated distances of approximately 80 Å, consistent with both C-FRET and 2p-FRET data from the proximal surface of 40-μm tissue Figure 5Multiphoton images from 6-h-postinjury tissue in 60- (a) to (c), 80- (d) to (f), or 100- (g) to (i) μm-thick tissue. Following immunohistochemical processing with mouse anti-BAD IgG 1 labeled with Alexa 488 anti-mouse antibody (donor) and/or rabbit anti-Bcl-xL IgG labeled with Alexa 555 anti-rabbit IgG (acceptor), images required for PFRET analysis were acquired. In 6-h-postinjury animals, axons demonstrated ubiquitous labeling for BAD with excitation with a multiphoton in all tissue thicknesses (a) 60, (d) 80, and (g) 100 μm, significant acceptor emission is detected with donor excitation in all thicknesses (b), (e) and (h), and PFRET image correction demonstrates energy transfer from donor to acceptor (c), (f), and (i) with mean efficiency and estimated distance in angstroms. Bar=10 μm. Color Plate 2  4.DiscussionFRET microscopy is a powerful tool for visualizing protein-protein interactions under physiological conditions. In this paper, we report a reliable and repeatable methodology for FRET microscopy employing conventional immunohistochemical approaches in fixed tissue. The development of a feasible method for employing FRET microscopy in fixed tissue with conventional immunohistochemistry constitutes a significant advance over prior preparatory approaches. Specifically, to this point, FRET microscopy has primarily been employed to explore protein interactions in vitro. In this setting, expression vectors are constructed containing proteins of interest adjacent to sequences encoding donor and acceptor fluorophores such as green fluorescent protein (GFP) and yellow fluorescent protein (YFP). The two vectors are introduced in vitro and expressed. As the proteins of interest interact, they reside within 10 to 100 Å of each other and radiationless interactions occur between donor and acceptor fluorophores that can be detected by FRET microscopy. This technique was employed to examine receptor-ligand interactions, including binding between the estrogen receptor α- and β-subunit and steroid receptor coactivators in the presence of estradiol.19 Similarly, ligand-independent ErbB1 receptor activation following focal application of EGF has also been assessed through this approach.20 In addition to ligand-dependent and -independent interactions, the creation of genetic constructs with adjacent fluorophores designed for FRET microscopy has been used to assess transient protein assemblies. An example includes receptor-mediated formation of the G protein heterotrimer through labeling of the α-subunit and the β-subunit.21 Further, Everett et al.;22 used this approach to demonstrate that the myxoma poxvirus protein M11L prevents apoptosis by interacting with the mitochondrial transition pore. FRET pair constructs have also been used to produce an assay to detect caspase activation.23 In addition to the design of expression vector constructs with respective donor and acceptor molecules adjacent to proteins of interest, immunocytochemical approaches have been employed using donor and acceptor fluorophores conjugated directly to primary antibodies.24 An example of how this approach has been utilized includes the characterization of epitope-specific anti-platelet antibodies.25 Further, Kam et al.;26 utilized this direct immunocytochemical methodology to map cytoskeletal components of adherens junctions by demonstrating interactions between vinculin and talin. Kenworthy et al.;27 used antibodies linked to GPI-anchored proteins to characterize plasma membrane microdomains. Bruno et al.;28 developed a highly sensitive assay for Bacillus spores and E. coli O157:H7 by measuring decreases in FRET activity. Additionally, Sharma et al.;29 used antibodies against Torsin A and α-Synuclein to demonstrate the association between these proteins in a form of inherited Parkinson’s disease. Both the design of constructs with fluorophores incorporated into fusion proteins and direct immunohistochemical approaches have yielded significant insights into a wide variety of protein interaction in relation to intact cells. However, the currently discussed technique for employing conventional immunocytochemical approaches in fixed tissue for FRET microscopy overcomes a number of limitations presented the previous approaches. Specifically, the construction and introduction of fusion proteins with incorporated fluorophores requires the adequate delivery, incorporation, and expression of donor and acceptor molecules in living systems prior to their characterization under FRET microscopy. In contrast, the currently described method of tissue FRET microscopy does not require the introduction or incorporation of any exogenous materials to cells or tissue while in the living state. Further, use of primary antibodies directly conjugated to fluorophores is a more wasteful and time-consuming process than using conventional immunohistochemical approaches with primary and secondary antibodies. Specifically, as primary antibodies are only rarely available directly conjugated to a fluorophore, a preliminary procedure must be carried out to conjugate a fluorophore to the primary antibody of interest. This procedure is not only time consuming, but also results in the wasting of a proportion of the primary antibody that is not conjugated to fluorophore as a function of the process. Further, using primary antibodies directly conjugated to fluorophores does not allow for the amplification of signal that is typically afforded by using primary and secondary antibodies, resulting in the necessity for higher initial concentrations of the primary antibody. Both the incomplete conjugation of primary antibody and the requisite for higher primary antibody concentrations are significant considerations in experiments where the primary antibodies may be rare and/or costly. An important element of the current approach is the use of pixel-by-pixel analysis of FRET images acquired by either C-FRET or 2p-FRET microscopy. The use of PFRET software to perform pixel-by-pixel analysis enables the quantitative detection and subtraction of DSBT and ASBT from FRET images while assessing the degree of FRET interaction. While FRET analysis can be conducted in tissue using photobleaching techniques to decrease the level of DSBT and ASBT (Ref. 29), pixel-by-pixel analysis has distinct advantages over photobleaching. Specifically, with pixel-by-pixel analysis the tissue specimen receives a low energy exposure during two brief sequential image acquisitions. Photobleaching instead requires an extended exposure to in the acceptor excitation spectral range to quench the acceptor molecule. In addition to the decreased ability to assess and subtract DSBT and ASBT in a quantitative and controlled fashion, we observed tissue alteration and destruction following exposure to extended and high excitation energy levels such as those associated with photobleaching. An additional element of the current communication is the use of 2p-FRET microscopy to localize the protein-protein interactions in tissue. While confocal microscopy provides a powerful tool for obtaining the images required for tissue FRET, 2p microscopy has several advantages. First among these is the reduced focal volume imaged in 2p microscopy. Confocal microscopy uses a pinhole to reduce the focal plane by blocking out of focus emissions; 2p microscopy instead accomplishes a decreased focal volume by directly reducing the excitation focal plane depth. A second advantage of 2p microscopy is that the use of lower frequency wavelengths to excite the fluorophores results in a lower energy load to the specimen. While this may not be of major importance in the fixed specimens used in this study, it may be a critical factor in studies involving living tissue. A third advantage of 2p microscopy is the increased ability to penetrate and visualize deeper tissue. In this experiment, we analyzed tissue up to 100 μm thick, obtaining good-quality images of individual axons, and were able to determine FRET efficiencies consistent with thinner tissue and with confocal data. In this paper, we report on the use of tissue FRET microscopy to evaluate the interactions between BAD and Bcl-xL in TAI following TBI. TAI is one of the most devastating components of TBI involving the widespread but diffuse disconnection of axons in the hours to days following a traumatic insult. One of the primary challenges to the exploration of TAI is the diffuse nature of the event. While the damage occurs at a broad enough scale to severely disrupt neurophysiological processes, the diffuse nature of the event, occurring directly adjacent to normal tissue, has primarily confined investigations on the pathogenesis of TAI to immunocytochemical approaches. A further limitation to the study of TAI is that investigations are optimally carried out in vivo. While there do exist in vitro models aimed at describing how stress and strain may affect individual axons and neuronal cell bodies following TBI, injury resulting from the complex biomechanical interactions between glia, neurons, axons, and the extracellular milleu cannot be adequately assessed in vitro. Our current hypothesis on the pathogenesis of TAI based on previous immunocytochemical and histochemical studies involves an initial loss of ionic homeostasis secondary to traumatically induced axolemmal perturbation. This loss of ionic homeostasis leads to activation of Ca 2+ -mediated second messenger processes,6 potentially including those mediated by Calmodulin. Calmodulin activates a number of targets including the phosphatase calcineurin, which can dephosphorylate and activate BAD, a proapoptotic protein. Dephosphorylated BAD travels to the mitochondria and binds to a prosurvival protein, Bcl-xL, leading to the breakdown of mitochondria.7 Mitochondrial breakdown and release of cytochrome c results in the activation of the caspase cascade of proteolytic enzymes, culminating in local axonal breakdown.8 9 Neither conventional immunocytochemical/histochemical nor ultrastructural approaches have the capacity to shed insight on whether BAD and Bcl-xL interact in TAI in vivo. The development of a tissue FRET technique enabled us to demonstrate BAD and Bcl-xL interaction while preserving the complex biomechanical interactions and tissue architecture between glia, neurons, axons, and the extracellular milleu. In this paper, we have demonstrated that following TAI, BAD and Bcl-xL interact in axons displaying the morphological characteristics of injury, thus suggesting the interaction between these two molecules may play an important role in TAI pathogenesis. AcknowledgmentsWe wish to thank Ms. Ye Chen for her valuable help in FRET data processing and the W. M. Keck Foundation and the Commonwealth Neurotrauma Initiative for their support. REFERENCES
R. J. Waxweiler
,
D. Thurman
,
J. Sniezek
,
D. Sosin
, and
J. O’Neil
,
“Monitoring the impact of traumatic brain injury: a review and update,”
J. Neurotraum. , 12
(4), 509
–516
(1995). Google Scholar
D. M. Sosin
,
J. E. Sniezek
, and
R. J. Waxweiler
,
“Trends in death associated with traumatic brain injury, 1979 through 1992. Success and failure,”
JAMA , 273
(22), 1778
–1780
(1995). Google Scholar
E. M. Doppenberg
,
S. C. Choi
, and
R. Bullock
,
“Clinical trials in traumatic brain injury. What can we learn from previous studies?,”
Ann. N.Y. Acad. Sci. , 825 305
–322
(1997). Google Scholar
R. Raghupathi
,
D. I. Graham
, and
T. K. McIntosh
,
“Apoptosis after traumatic brain injury,”
J. Neurotraum. , 17
(10), 927
–938
(2000). Google Scholar
A. M. Petros
,
D. G. Nettesheim
,
Y. Wang
,
E. T. Olejniczak
,
R. P. Meadows
,
J. Mack
,
K. Swift
,
E. D. Matayoshi
,
H. Zhang
,
C. B. Thompson
, and
S. W. Fesik
,
“Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies,”
Protein Sci. , 9
(12), 2528
–2534
(2000). Google Scholar
H. G. Wang
,
N. Pathan
,
I. M. Ethell
,
S. Krajewski
,
Y. Yamaguchi
,
F. Shibasaki
,
F. McKeon
,
T. Bobo
,
T. F. Franke
, and
J. C. Reed
,
“
Ca2+
-induced apoptosis through calcineurin dephosphorylation of BAD,”
Science , 284
(5412), 339
–343
(1999). Google Scholar
J. E. Springer
,
R. D. Azbill
,
S. A. Nottingham
, and
S. E. Kennedy
,
“Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury,”
J. Neurosci. , 20
(19), 7246
–7251
(2000). Google Scholar
A. Buki
,
D. O. Okonkwo
,
K. K. Wang
, and
J. T. Povlishock
,
“Cytochrome c release and caspase activation in traumatic axonal injury,”
J. Neurosci. , 20
(8), 2825
–2834
(2000). Google Scholar
B. A. Eldadah
and
A. I. Faden
,
“Caspase pathways, neuronal apoptosis, and CNS injury,”
J. Neurotraum. , 17
(10), 811
–829
(2000). Google Scholar
R. N. Day
,
“Visualization of Pit-1 transcription factor interactions in the living cell nucleus by fluorescence resonance energy transfer microscopy,”
Mol. Endocrinol. , 12
(9), 1410
–1419
(1998). Google Scholar
A. Periasamy
,
“Fluorescence resonance energy transfer microscopy: a mini review,”
J. Biomed. Opt. , 6
(3), 287
–291
(2001). Google Scholar
R. B. Sekar
and
A. Periasamy
,
“Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations,”
J. Cell Biol. , 160
(5), 629
–633
(2003). Google Scholar
A. Marmarou
,
M. A. Foda
,
B. W. van den
,
J. Campbell
,
H. Kita
, and
K. Demetriadou
,
“A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics,”
J. Neurosurg. , 80
(2), 291
–300
(1994). Google Scholar
J. R. Stone
,
S. A. Walker
, and
J. T. Povlishock
,
“The visualization of a new class of traumatically injured axons through the use of a modified method of microwave antigen retrieval,”
Acta Neuropathol. (Berl.) , 97
(4), 335
–345
(1999). Google Scholar
M. Elangovan
,
H. Wallrabe
,
Y. Chen
,
R. N. Day
,
M. Barroso
, and
A. Periasamy
,
“Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy,”
Methods , 29
(1), 58
–73
(2003). Google Scholar
W. Denk
,
J. H. Strickler
, and
W. W. Webb
,
“Two-photon laser scanning fluorescence microscopy,”
Science , 248
(4951), 73
–76
(1990). Google Scholar
A. Periasamy
,
P. Skoglund
,
C. Noakes
, and
R. Keller
,
“An evaluation of two-photon excitation versus confocal and digital deconvolution fluorescence microscopy imaging in Xenopus morphogenesis,”
Microsc. Res. Tech. , 47
(3), 172
–181
(1999). Google Scholar
Y. Bai
and
V. Giguere
,
“Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells,”
Mol. Endocrinol. , 17
(4), 589
–599
(2003). Google Scholar
P. J. Verveer
,
F. S. Wouters
,
A. R. Reynolds
, and
P. I. Bastiaens
,
“Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane,”
Science , 290
(5496), 1567
–1570
(2000). Google Scholar
C. Janetopoulos
,
T. Jin
, and
P. Devreotes
,
“Receptor-mediated activation of heterotrimeric G-proteins in living cells,”
Science , 291
(5512), 2408
–2411
(2001). Google Scholar
H. Everett
,
M. Barry
,
X. Sun
,
S. F. Lee
,
C. Frantz
,
L. G. Berthiaume
,
G. McFadden
, and
R. C. Bleackley
,
“The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore,”
J. Exp. Med. , 196
(9), 1127
–1139
(2002). Google Scholar
J. Jones
,
R. Heim
,
E. Hare
,
J. Stack
, and
B. A. Pollok
,
“Development and application of a GFP-FRET intracellular caspase assay for drug screening,”
J. Biomol. Screen. , 5
(5), 307
–318
(2000). Google Scholar
A. K. Kenworthy
,
“Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy,”
Methods , 24
(3), 289
–296
(2001). Google Scholar
M. Koksch
,
G. Rothe
,
V. Kiefel
, and
G. Schmitz
,
“Fluorescence resonance energy transfer as a new method for the epitope-specific characterization of antiplatelet antibodies,”
J. Immunol. Meth. , 187
(1), 53
–67
(1995). Google Scholar
Z. Kam
,
T. Volberg
, and
B. Geiger
,
“Mapping of adherens junction components using microscopic resonance energy transfer imaging,”
J. Cell. Sci. , 108 1051
–1062
(1995). Google Scholar
A. K. Kenworthy
,
N. Petranova
, and
M. Edidin
,
“High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes,”
Mol. Biol. Cell , 11
(5), 1645
–1655
(2000). Google Scholar
J. G. Bruno
,
S. J. Ulvick
,
G. L. Uzzell
,
J. S. Tabb
,
E. R. Valdes
, and
C. A. Batt
,
“Novel immuno-FRET assay method for Bacillus spores and Escherichia coli O157:H7,”
Biochem. Biophys. Res. Commun. , 287
(4), 875
–880
(2001). Google Scholar
N. Sharma
,
J. Hewett
,
L. J. Ozelius
,
V. Ramesh
,
P. J. McLean
,
X. O. Breakefield
, and
B. T. Hyman
,
“A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study,”
Am. J. Pathol. , 159
(1), 339
–344
(2001). Google Scholar
|


CITATIONS
Cited by 48 scholarly publications.
Tissues
Fluorescence resonance energy transfer
Microscopy
Axons
Proteins
Confocal microscopy
Injuries


