
|
|
1.IntroductionFörster resonance energy transfer (FRET),1 which relies on nonradiative quenching of intensity and lifetime of a donor fluorophore to an acceptor, is useful due to its length scale (1– ) being similar to those of interest in cell biochemistry. Techniques for lifetime measurement fall into two categories:2 the frequency domain in which the sample is excited with sinusoidally modulating light, with the optimal angular frequency being the reciprocal of the lifetime;3, 4 and time domain, whereby the fluorophore is excited by an ultrafast light pulse and fluorescence lifetime is directly measured from the decay using time-correlated single photon counting (TCSPC).5 The advantage of the time-domain technique is the comparative ease by which multiple lifetimes and complex decay profiles can be extracted using only a single excitation wavelength.6 This allows assessment, simultaneously, of both the percentage of interacting molecules as well as the proximity of the interaction. The in vitro study of protein interactions and tertiary conformation using FLIM with sufficient signal to noise to allow the separation of multiple exponentials in single cells requires approximately of acquisition time per cell. There has been a large investment in high-throughput screening technology and techniques over the past decade. High-throughput frequency domain fluorescence plate readers and times-resolved energy transfer (TRET) assays, using 96-well microtitre plates,7, 8 are currently being used. There have been recent developments in wide field time-domain FLIM techniques using microtitre plates assuming a simple mono-exponential decay function.9 A time-domain fluorescent lifetime plate reader has not yet been demonstrated to be useful for the measurement of complex decay profiles expected from protein–protein interaction or conformational assays and are not yet applied to drug discovery. Here we evaluate a time-domain fluorescent lifetime plate reader. We demonstrate the necessity of eliminating the complex background autofluorescence from the signal for accurate measurement and describe a custom-written analysis program that effectively separates multiple lifetimes from the background. We make use of the stretched exponential function (StrEF), which has been used to describe decay phenomenon with distributions of lifetimes and as an approximation to multiple exponential decay profiles.10, 6 We go on to demonstrate the successful application of the instrument to both detect protein–protein interactions and perform a protein conformational assay using a cell-based format. As a practical example of this approach, we apply the technique to a problem derived from Alzheimer disease research. Extracellular plaques comprised primarily of Amyloid- (A ) peptide are a major neuropathological hallmark of Alzheimer’s disease (AD). Proteolysis of A involves cleavage of amyloid precursor protein (APP) by - and -secretase complexes, the latter of which is Presenilin-1-(PS1) dependent.11, 12, 13 PS1 is a 467-amino acid, 9-transmembrane domain protein. Over 100 documented single-point mutations in the gene that expresses the protein are known to cause autosomal dominant familial AD (FAD),14 in which levels of A are increased and the fraction of the longer, more fibrillogenic form of the peptide (A -42) is increased.15 We apply the plate reader and our analysis technique to a recently developed assay that reflects the conformation of PS1 in intact cells.16, 17 We also demonstrate that the plate-reader-based technique is sensitive to changes in PS1 conformation between a mutant C410Y variant known to cause FAD and the wild-type (WT) protein. PS1 is a multipass transmembrane protein that is a component of a large -secretase complex, responsible for cleavage of the amyloid precursor protein and multiple other substrates. 2.Materials and Methods2.1.InstrumentWe used a multifunction microplate detector (UltraEvolution FLT, Tecan Trading, Switzerland) for this study. The instrument consists of a high-throughput optical detector with two picosecond-pulsed semiconductor lasers at and . Light is detected with a photo-multiplier tube (PMT), connected to a time-correlated single-photon counting (TCSPC) board (Picoquant, GmBH). Light ( for all experiments described here) is conveyed via optical fibers to a series of Neutral Density 2 mirrors on a translation stage, allowing for the brightness of the laser pulse to be attenuated to avoid photon pileup,5 before being reflected into the sample. Light emitted from the sample is passed through an interchangeable emission filter ( for all studies performed here) before being transmitted to the detector. The instrument initially had light collection optics for both transmission and reflection geometry. The bottom-mounted transmission optics were replaced with a beam block (Thorlabs, NJ) to reduce reflection and autofluorescence. For all measurements, 96-well, black, untreated glass-bottom plates (Nalge NUNC International, Rochester, NY) were used, as they were found to have low autofluorescence. 2.2.Fluorescent DyesAlexafluor 430 (A430) (Invitrogen, CA)18 was chosen as a suitable fluorescent dye because the peak of its excitation spectrum is close to the wavelength of one of the instrument lasers. It also has a large stokes shift with the peak of the emission spectrum being and overlapping well with the excitation spectrum of Cyananine3 (cy3) dye (Invitrogen, CA), which was chosen as a suitable acceptor. 2.3.Cell PreparationsThree cell lines were used during this investigation: Chinese hamster ovary (CHO); PS70, CHO cells stably transfected with wild-type (WT) PS1 and WT amyloid precursor protein 770 (APP); and C410Y, CHO cells stably transfected with Familial Alzheimer’s disease associated mutant PS1 and WT APP.17 Cells were grown in Opti-MEM (Gibco) supplemented with 10% feotal bovine serum, G418, and puromycin at with 5% . For experiments, the PS70 cells were split onto black 96-well plates coated with poly L-lysine. The cells were allowed to grow to confluency prior to immunocytochemistry. Cells were fixed in methanol ( , ), permeabilized, and nonspecific binding sites blocked (5% normal donkey serum, 0.1% Triton-X 100, ). To create a model cell-based FRET assay, cells were immunostained with a mouse antitubulin primary antibody (Abcam, Cambridge, MA) overnight at , and goat antimouse secondary antibody, labeled with A430 (1:100) (Invitrogen, Carlsbad, CA), at room temperature. Cells in some wells were additionally stained with a donkey antigoat tertiary antibody labeled with Cy3 (1:200) (Jackson Immuno Research) for at room temperature. This protocol creates a condition whereby A430 (donor) is brought into close proximity to Cy3 (acceptor) so that FRET can occur. Controls included unstained cells as well as cells immunostained with the acceptor or donor alone. PS70 and C410Y cells were used in a demonstration of a conformation assay.19 Termini of PS1 were immunostained with rabbit -human S182 antibody against amino acids 450–467 (C-terminus) (Sigma, St Louis, MO) and goat -human PS1 directed against amino acids 14–33 (N-terminus) (Sigma). Cells were subsequently incubated with goat -rabbit A430 and donkey antigoat Cy3 ( , room temperature). Alternatively, cells were immunostained with C8 antibody raised against amino acids 676–695 of APP (20) and an antibody to the major loop between PS1 TM6 and TM7 (mouse -PS1, amino acids 263–378; Chemicon, Temecula, CA). Secondary antibodies were goat -mouse A430 and donkey -rabbit Cy3. In all preparations, cells were washed three times in 1X TBS between and after rounds of staining. 2.4.Curve FittingDuring FRET,21 energy is nonradiatively transferred from a donor fluorophore to an acceptor molecule. Although the transfer is nonradiative, the emission spectrum of the donor must overlap with the excitation spectrum of the acceptor. It is not necessary for the acceptor to be a fluorophore, although they are generally used. The effect of the transfer is to excite (increase the brightness) of the acceptor population while quenching (decreases in both brightness and lifetime) the donor. The stretched exponential function10 (StrEF) differs from the simple exponential (SimEF) by the inclusion of an extra characteristic constant, , which represents the degree of stretch of the function, We suggest the use of it here to describe the background autofluorescence of the instrument and sample assay as it can adequately describe a broad range of continuous and discrete multi-exponential functions.6 From a practical perspective, the simplicity and generality of the StrEF, compared to a complex multi-exponential function, make it easier to apply. This is especially important in a high-throughput or laboratory environment where various conditions, antibodies, or cell types may be used, provided that the StrEF adequately describes the background autofluorescence and allows discrimination of FRETing and non-FRETing conditions. A custom-written analysis named General Multi-Exponential Fitter (GeMEF) was created using Matlab (Mathworks, Novi, MI). We used a generalized multi-exponential function, which can represent a sum of StrEF or SimEF functions: where represents measured intensity at time , is a constant offset, is the fractional contribution of each component and so is the contribution of each component at is the characteristic lifetime of each component, and is the degree of stretch. The equation describes the decay curve of fluorophore populations. The basis of the algorithm is a nonlinear, maximum-likelihood estimation (MLE), using the Matlab function fminsearch, which makes use of an unconstrained direct search method (Nelder–Mead). Points were weighted according to their estimated noise assuming Poisson statistics [ , deviation]. The MLE has been shown to be more accurate and less dependent on measurement noise than the least-squares (LS) method.22 Each term in the equation can be set to a constant value or allowed to be fit, depending upon requirements.2.5.Calculation of FRET Strength and EfficiencyThe number of molecules of a given population is proportional ; the proportion of the donor molecules that are interacting (FRET strength) is given by where the subscripts 1 and 2 refer to the non-FRETing and FRETing populations of the donor fluorophore, respectively. The fraction of energy transferred during the interaction FRET efficiency is related to the distance between the two fluorophores and is given by23 where represents the distance between the donor and acceptor fluorophore and refers to the Förster distance—the distance at which 50% of the donor’s excitation energy is transferred to the acceptor.2.6.Experimental ProtocolsFor initial characterization of the noise distribution and background autofluorescence, wells containing PBS (pH 7.4) were fit with parameters , , , , and allowed to vary while the other parameters are fixed with all other values of . We use the subscript to refer to background. The sensitivity of the instrument to dye concentration was assessed by varying concentrations of free A430 ( in pH 7.4 PBS) (eight wells per condition, spread over two 96-well plates). A set of eight wells per plate contained saline alone. The level of attenuation of the excitation light was tuned to maximize photon count while avoiding pileup. For low concentrations of dye, when the laser was not strongly attenuated, the background autofluorescence was measured from the wells with no fluorophore and those parameters were fixed before fitting a mono-exponential curve to the data. In the case where laser power was significantly attenuated, the background autofluorescence was too weak to accurately measure. Under those conditions, given the relative strength of the signal, the background was assumed insignificant and ignored. A series of conditions with varying FRET strengths but the same FRET efficiency were made from solutions of goat antimouse primary antibody , labeled with A430,18 which were incubated with varying amounts of biotinylated (nonfluorescent) antigoat (4 – ) secondary antibody in of PBS, for at room temperature. Donkey antigoat, labeled with Cyanine 3 (Cy3) , was added and incubated for a further hour at room temperature. The solutions were transferred to a 96-well plate. Finally, CHO, PS70, and C410Y cells were grown in 96-well plates and immunolabelled as described above. For the model FRET assay experiment, eight wells were used for both negative controls and experimental samples. For the PS1 conformation and PS1-APP studies, 16 wells were used per condition and for negative controls. Eight wells containing unstained cells were used for background assessment. The background assessment was conducted as described above. The lifetime of the non-FRETing donor fluorophore was then obtained by examining wells containing the donor but no acceptor. During this stage, a second exponential term was introduced and values of were fit; was fixed to 1. Finally, the lifetime of the FRETing donor population and relative amplitudes of the non-FRETing and FRETing populations were obtained from wells containing the complete assay, while was fixed at the previously obtained value and was fixed to 1; the nonacceptor wells were fit again under these conditions, as negative controls. 3.Results3.1.Background Autofluorescence and Noise CharacteristicsWells in a 96-well plate that contain of saline were used to assess the signal due to instrument and plate autofluorescence and the validity of the noise model (Fig. 1 ). The goodness-of-fit measure, reduced for the SimEF, was 2.86. The time course of the residuals, divided by the expected standard deviation according to Poisson statistics , corrects for the time dependency of the noise model. The residuals for the SimEF are not normally distributed around zero at all points during the decay. For the stretched exponential, is achieved and the residuals appear to be more normal. Fig. 1Analysis of background fluorescence. A typical time-resolved fluorescence signal from a Nunc 96-well, glass-bottomed plate containing of saline is shown fitted with both simple A and stretched B exponential curves. The residuals are also shown, normalized by expected noise . Panels C and D show the normalized residual distributions fitted by a Gaussian. The width of the Gaussian and the regression value are shown. The autocorrelations (AC) (panels E, F) and the goodness of fit to of all coefficients apart from the central value .  Two further tests of goodness of fit were performed to quantify the improvement to fit of using the StrEF. Fitting the distribution of the corrected residuals to a Gaussian gives an indication of how well the residuals conform to the normal distribution (Figs. 1C+D).24 Ideally, the width of the fitted Gaussian and the goodness-of-fit measure should both be unity. The stretched exponential conforms better to the Gaussian ( compared to for the simple exponential). The standard deviation obtained from the width of the fitted Gaussian is close to 1 for the StrEF but larger for the simple exponential . The second test was to check for the statistical independence of various time points using the autocorrelation (AC) function24 of the residuals , where is the temporal shift. For the SimEF (Panel C), strong coefficients are found at shifts other than 0. The goodness of fit to for all nonzero lags is shown to be . For the StrEF, the AC function is much flatter for all nonzero shifts and ; this shows that the residuals are less correlated (whiter) for the StrEF. Although these data partially validate the use of the StrEF, for complete validation, the technique must be applied to a model protein–protein interaction to prove its ability to discriminate between positive and negative FRET conditions. Simulations to study the effect of noise level on the fitting parameters were conducted by generating time courses of the form where is Poisson noise. The results of fits to these simulations were compared to the variations observed from a reading of a 96-well plate (Table 1 ). The variation in amplitude of the background function ( since and there are no other components) is approximately a factor of 9 more than expected from simulation, while for and , the figure is around 4. The comparison shows that there are other significant sources of error than can be accounted for by measurement noise. For this reason, simulated errors were not relied upon as an indication of uncertainty, error bars were defined by the standard deviations of measured results.Table 1Comparison of Standard Deviations in Fitting Parameters Expected from Poisson Statistics and Measured Standard Deviations (n=96) .
3.2.Assessment of Instrument Sensitivity to Fluorophore ConcentrationVarying concentrations of free A430 were used to test the sensitivity of the instrument. At the greatest laser attenuation (ND1024), the background autofluorescence was negligible. Decay curves were fit with a mono-exponential plus an offset. The amplitude (Fig. 2A ) varies linearly with concentration. The lifetime ( , Fig. 2B) is stable over three orders of magnitude of concentration at . At . Concentrations higher than could not be analyzed due to photon pile-up; at lower concentrations, the number of photons was too small to fit. At minimum laser attenuation (NDI), the background was fitted with a stretched exponential and subtracted from the signal. The intensity varied linearly with concentration (Fig. 2C). At , observed at . At lower concentrations, both and increase as the signal level decreases. At the minimum measurable concentration , the value of is 0.14% higher than the value given at high concentrations (Fig. 2D). At concentrations below , the signal was too weak to fit, causing large variations in fitted values (data are not shown). These data show that amplitude varies linearly with concentrations over 4 orders of magnitude while remains almost completely stable. Even at low signal levels, the fitted lifetime is remarkably close to the results taken at high concentrations. Fig. 2Measured fluorescence signal rises monotonically with an increased fluorophore concentration. At higher laser attenuation (Panel A), the background autofluorescence was not subtracted and intensity varied linearly over 4 orders of magnitude. The lifetime (Panel B) was found to be very stable although it was found to be slightly lower with greater error at the lowest concentration. At a lower attenuation, the background autofluorescence was subtracted. The intensity was found to be linearly dependent on concentration (Panel C). The fitted lifetime did not vary significantly; the standard deviation increased at low concentrations but was small (Panel D). The amplitude of the fitted curve is shown to vary monotonically with respect to molarity. The background subtraction was performed on the data in Panels C and D. Lifetimes were found to be concentration-independent at standard deviation, for all fits. 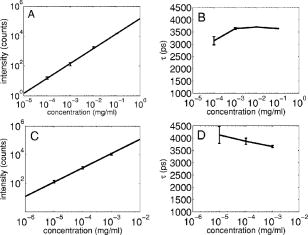 3.3.Simulations of the Effect of Background Autofluoresence on MeasurementTo assess the impact of background autofluorescence as a confound and show that it is necessary to subtract it before fitting, a mono-exponential decay was simulated and analyzed using the standard bi-exponential model used in most FLIM experiments. The sum of a stretched exponential with parameters similar to the background function observed and a mono-exponential ( , where the subscript means simulated) with varying relative amplitudes were fit with a bi-exponential using GeMEF. A time base similar to that generated by the instrument ( points) was used. The relative amplitude (Fig. 3A ) of the component with the shorter lifetime rises as the signal strength increases. The shorter lifetime is closer to and lower than the simulated lifetime. It has a minimum where the signal strength is about twice the background, while increases monotonically with signal strength. Fig. 3The extent to which a stretched exponential background would lead to an apparent second exponential in the analyzed data. A stretched exponential function similar to the one observed in the instrument is added to a single exponential of varying intensities. Panels A and B show the relative amplitude of the shorter lifetime term and the returned lifetime against signal strength normalized by the amplitude of the simulated background function. Panels C and D show the case in which an offset was added, for which the fitting routine fit. The results show that the contribution of the stretched exponential background function would result in two apparent decay components and therefore false positive FRET results. 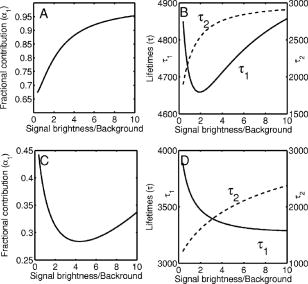 An offset might compensate for a long-lived component. A constant was added and the fitting routine was allowed to fit it. As the signal strength increases, the two lifetimes converge toward . The longer-lived component is stronger but does not dominate (Fig. 3C). In both simulations, a significant false positive result was predicted when background autofluorescence was not accounted for, especially at the low signal strengths expected from cell-based assays. 3.4.Measurement of FRET Strength at Constant FRET EfficiencyAn important requirement of time-domain FRET data analysis is the ability to distinguish between the parameters of FRET strength and efficiency. Here we determine whether our calculations effectively distinguish these two parameters with minimal cross-talk. Donor-labeled antibody was pre-incubated with varying amounts of biotinylated secondary antibody to reduce the number of available epitopes, before being incubated with acceptor-labeled secondary. The proportion of primary antibody that is bound to secondary labeled with acceptor should be inversely proportional to the concentration of the biotinylated secondary. The inverse of the FRET strength increases linearly with no change in FRET efficiency (Fig. 4 ). These data illustrate our ability to distinguish between a change in the proportion of proteins interacting and the closeness with which they interact. Fig. 4Our analysis separates FRET strength and efficiency with minimal cross-talk. Goat antimouse primary antibody, pre-incubated for with varying amounts of donkey biotin antigoat was incubated for a further hour with Cy3 labeled donkey antigoat antibody. The biotin-labeled secondary blocks the binding sites available to the labeled secondary, leading to a reduction in FRET strength (Panel A),without changing FRET efficiency (Panel B).  3.5.Model Protein–Protein Interaction AssayMeasurements of protein interactions in cell systems using immunohistochemistry or fluorescently labeled protein expression are an important experimental use of plate readers. Here we demonstrate the use of the plate reader to detect a protein–protein interaction and show the importance of subtracting the background autofluorescence to avoid false positive results. Sufficient signal was detected from a monolayer of adherent cells immunostained with A430, using tubulin as the antigen, in the 96-well format with the instrument. The lifetime of A430 was found to be (averaged over eight wells). Controls included wells treated with either the primary or secondary antibodies alone, from which the signal was negligible. The model protein–protein interaction described in the methods produces a circumstance in which tubulin is labeled with both A430 and Cy3, bringing the A430 and Cy3 into close proximity. The control for this experiment involved the tertiary antibody conjugated to unlabeled biotin. FRET strength was used as a measure of the presence of a protein–protein interaction. For the negative control, FRET strength was not significantly larger than zero (Fig. 5A ) . For the double stained cells, FRET strength was 0.81. The lifetime of the second component was found to be . If the background autofluorescence was not subtracted, a significant false positive FRET strength was observed, (Fig. 5B, ). The FRET strength in the model interaction dropped to 0.7414. The false positive result here illustrates the value of background correction in circumstances such as cell-based assays, in which the amount of available signal is limited compared to the background auto-fluorescence. Furthermore, the use of the StrEF is validated since it leads to the elimination of false positive results. Fig. 5Comparison of apparent FRET strength for a model protein–protein interaction, in a cell-based assay, compared to a negative control. Tubulin was immunostained with A430 and either Cy3 or biotin-labeled secondary antibodies. A student’s t-test of the amplitude of the negative control shows that is not significantly greater than zero (Panel A). For comparison, the results from a simple bi-exponential fit without background correction is shown (Panel B). A significant false positive result is obtained if the background is not subtracted . 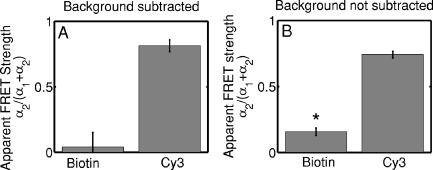 3.6.Analysis of Protein Conformation: Alzheimer’s Disease Associated with Presenilin-1 ProteinThe question of PS1 conformation has been studied using time-domain FLIM microscopy by comparing apparent lifetime changes of donor fluorophores.17 Because it is a multipass transmembrane protein, its structure has been difficult to assess; a recent fluorescence lifetime microscopy study, however, showed that antibodies directed at dual epitopes of PS1 change their relative proximity to each other in mutant, compared to WT PS1.17 We show that our current plate reader-based analysis techniques allow us to detect this subtle change. For the negative control (Cy3 was omitted), was not statistically higher than zero ( wells), showing that there was no confounding second lifetime due to local variations in the cellular environment. The C- and N-termini of PS1 in the mutant cell line (C410Y) showed significantly stronger FRET than the wild-type (PS70) ( , wells per condition) (Fig. 6A ). A comparison of FRET efficiency shows that the interterminal distance is less in the mutant C410Y variant of PS1 (Fig. 6B). These data confirm previous work that the mutant variety of PS1 has a measurable conformational difference with the C- and N-termini being closer together. Fig. 6Results of protein conformation assay of C- and N-terminal FRETing interaction. CHO cells were stably transfected with either mutant PS1 (C410Y cell line), known to cause FAD, or WT(PS70 cell line), and human APP. The C- and N-termini of PS1 were immunolabelled with A430 and Cy3. The mutant PS1 showed statistically significantly smaller C- and N-terminal distance (Panel A). There is also a significantly increased FRET strength (Panel B).  3.7.Analysis of Protein–Protein Interaction: Alzheimer’s Disease Associated with Presinilin-1 and Amyloid Precursor ProteinBerezovska also addressed the question of differences in the PS1-APP interaction between WT (PS70) and mutant (C410Y) forms of PS1. Here we test our ability to distinguish whether differences in FRET are due to closeness of the two fluorophores, which would indicate a difference in the PS1-APP closeness, or differences in the number of molecules interacting, which would indicate increased binding of PS1 to APP. We show here that using this instrument, a difference in FRET strength was detected (Fig. 7A ), which describes the proportion of donor molecules interacting with the acceptor, implying that the distance between the two proteins, when bound, is the same for both species of PS1 but that mutant PS1 has a greater propensity for binding APP. We can detect no statistically significant difference in the PS1-APP distance between the WT and mutant variants of PS1 (Fig. 7B). In addition, we notice that the FRET strength for both cell types in this experiment is particularly low . These data demonstrate that this assay can detect protein–protein interactions in intact cells. Fig. 7PS1-APP protein interaction assay. CHO cells stably transfixed with mutant or WT PS1 and human APP. PS1 was immunolabelled with A430 (donor) and APP was labeled with Cy3 (acceptor). There was a difference in FRETing strength (Panel A), but no statistical difference in PS1-APP distance between the WT and mutant varieties of PS1(Panel B). This result implies that there is greater interaction between mutant PS1 and APP but that the distance between PS1 and APP is similar between the mutant and WT PS1 conditions. 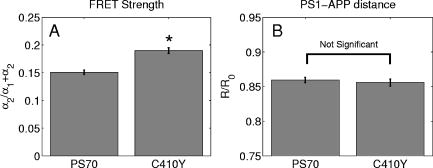 4.DiscussionWe have developed an analysis technique for FRET detection using a high-throughput time-domain fluorescence plate reader. We used a stretched exponential (StrEF) function as a general equation for the background autofluorescence of the plate and assay and demonstrate that it is necessary to remove the background to avoid false positive results. The seminal paper by Kohlrausch10 describes the decay of the residual charge on a glass Lyden jar with and shows that the function can be derived from an exponential with continuously increasing decay rate.25 More recently, numerous physical models of the underlying physics of StrEF in fluorescent and luminescent materials, involving progressively depleting random sinks that capture excitations,26 have been suggested, including, for example, fluorescent relaxation in glass.27 The StrEF has also been applied to biological samples.28 Here we use the StrEF as a generalized decay model to describe a series of nonspecific sources of background autofluorescence. We characterize the background autofluorescence from a 96-well plate containing only saline using the StrEF model and find that the model fits well to the data with the residual noise appearing to conform to a normal distribution and being white with respect to time, which validates our use of the StrEF. When comparing the results to a numerical simulation of interwell variance assuming Poisson statistics, we find a larger variance than we expect from measurement noise. The most likely explanation for this extra variance would be variations in the optical properties of the plates in which the measurement samples were placed, although variations in laser pulse characteristics cannot be ruled out. The errors shown here are small and not expected to impact the measurement results although even-greater sensitivity could be achieved with this instrument if sources of background autofluorescence were further reduced and optical properties of the sample plates could be made more uniform. Measurement of protein–protein interactions and protein conformations within fixed cells using immunochemistry and in living cells using fluorescently labeled proteins are important biological applications for plate readers, with a role in drug discovery and basic research into disease mechanisms. Time-domain lifetime detection is a particularly promising field as it allows for the measurement and diagnosis of complex decay profiles with high inherent accuracy and sensitivity. In principle, the approach is more powerful than spectral or time-resolved FRET techniques because it is concentration-independent, and allows simultaneous measurement of FRETing and non-FRETing populations as well as proximity measurements useful for monitoring conformational changes. Despite this, time-domain-based lifetime plate readers have not been used for FRET detection partly because of the confounds provided by complex decay profiles of background autofluorescence. Using a custom-written analysis program (GeMEF), we demonstrate the efficacy of this reader in a model system for studying protein–protein interactions in cells, by double immunostaining tubulin with donor and acceptor fluorophores. We present the analysis program (GeMEF), which progressively fits components of the decay curve and allows their separation. By comparing our corrected data to that generated by a simple bi-exponential fit, we demonstrate that it is necessary to fit to and remove background autofluorescence in order to avoid false positive results. As an example of the utility of the approach, we study a major target of pharmacological studies. PS1 is known to be a critical component of the -secretase complex that performs the final part of the proteolytic processing of APP to form A via intramembranous cleavage. PS1, a polytopic protein, must be post-translationally cleaved and associated with at least three other membrane-bound proteins to form a functional proteolytic complex. It has been previously demonstrated that point mutations in the PS1 protein alter the protein conformation and/or binding properties to APP.16, 17 We confirm previous microscope-based FLIM results showing that the conformation of a mutant form of PS1 (C410Y, FAD) is different, with the C- and N-termini being closer together, than the WT. We also measured the FRET efficiency in the condition where PS1 was immunolabelled with a donor fluorophore and APP was labeled with an acceptor. The current assay did not detect a previously noted subtle difference in PS1 to APP proximity between the two forms of PS1, but we did find that FRET strength was increased in the cells containing the mutant version of PS1. This observation suggests the possibility that one consequence of PS1 mutations is to change the properties of PS1 and its substrate, APP, consistent with the well-known consequence that PS1 mutants lead to a two-amino acid shift in the cleavage site of -secretase action on APP to generate A 42 to a greater extent than A 40.14 These results demonstrate the capacity of a plate-reader format assay to detect and measure protein conformation changes and changes in protein–protein interactions in cell-based formats, providing high-throughput data comparable to previous microscopy-based measurements.17 The previous work was performed using an FLIM microscopy technique, where many pixels were individually fit with bi-exponential curves and the lifetimes of a region of interest, representing a cell, were averaged. This process was repeated for many cells and average lifetimes were calculated. In practice, hundreds of cells must be individually measured for fluorescence lifetime imaging experiments, and each measurement takes on the order of . Our technique uses a single pixel covering many cells and much longer data acquisition times per pixel. As a result, the signal to noise using the new technique is significantly improved, potentially leading to improved separation of changes in lifetime from comparative amplitude of multiple decays. An entire 96-well plate can be read in less than at the expense of spatial information. The two techniques have the potential to be highly synergistic. For example, the observation that FRET strengths are low in the PS1-APP experiment may be partially explained by observations from the previous microscopy study,17 showing that both PS1 and APP are expressed throughout the cells but only interact at the membrane. Here we demonstrate, for the first time, a method of extracting complex decay profiles from time-domain lifetime assays in spite of the confound of complex decay profiles in the autofluorescent background. This technique is potentially extremely powerful as both a companion or a replacement technique for the process of single cell-based FLIM studies in the biological sciences but also as a high-throughput, high-accuracy screening technique for in vitro drug screening experiments. It should be noted, of course, that the same assays are even more powerful when measured in homogeneous solution, where the signal can be substantially higher than in intact cells. 5.ConclusionsWe have characterized the noise and sensitivity properties of a time-domain fluorescent lifetime plate reader and demonstrated its use for diagnosis of complex decay profiles. We go on to show the importance of subtracting the time-dependent background autofluorescent function and then present a custom-written analysis program (GeMEF) for the purpose of both removing background and progressively fitting lifetime components. Our analysis technique can effectively distinguish FRETing and non-FRETing populations and distinguish differences in FRET strength and efficiency. The GeMEF analysis is a potentially powerful tool for the study of protein–protein interactions, protein conformational studies, and drug screening experiments. We apply our technique to a previously examined assay of PS1 conformation and PS1-APP interaction in a cell-based format and demonstrate a remarkable increase in rapidity of generating data compared to existing microscope-based FLIM methods. AcknowledgmentsThe authors acknowledge support from the National Institutes of Health (NIH1R01EB00768, NIH2R01NF10828-30, NIH1T0AG15379-08). In addition we would like to thank David Boas and Sol Diamond for helpful discussions. ReferencesP. R. Selvin,
“The renaissance of fluorescence resonance energy transfer,”
Nat. Struct. Biol., 7
(9), 730
–734
(2000). https://doi.org/10.1038/78948 1072-8368 Google Scholar
P. I. H. Bastiaens and
A. Squire,
“Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell,”
Trends Cell Biol., 9
(2), 48
–52
(1999). https://doi.org/10.1016/S0962-8924(98)01410-X 0962-8924 Google Scholar
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd ed.Plenum Press, New York (1983). Google Scholar
J. R. Lakowicz and
B. P. Maliwal,
“Construction and performance of a variable-frequency phase-modulation fluorometer,”
Biophys. Chem., 21
(1), 61
–78
(1985). https://doi.org/10.1016/0301-4622(85)85007-9 0301-4622 Google Scholar
W. Becker,
A. Bergmann,
M. Hink,
K. König,
K. Benndorf, and
C. Biskup,
“Fluorescence lifetime imaging by time-correlated single-photon counting,”
Microsc. Res. Tech., 63
(1), 58
–66
(2004). https://doi.org/10.1002/jemt.10421 1059-910X Google Scholar
J. Siegel,
D. S. Elson,
S. E. Webb,
K. C. Lee,
A. Vlandas,
G. L. Gambaruto,
S. Leveque-Fort,
M. J. Lever,
P. J. Tadrous,
G. W. Stamp,
A. L. Wallace,
A. Sandison,
T. F. Watson,
F. Alvarez, and
P. M. French,
“Studying biological tissue with fluorescence lifetime imaging: Microscopy, endoscopy, and complex decay profiles,”
Appl. Opt., 42
(16), 2995
–3004
(2003). 0003-6935 Google Scholar
K. M. Swift and
E. D. Matayoshi,
“High throughput screening using dynamic fluorescence,”
Proc. SPIE, 2388 182
–188
(1995). https://doi.org/10.1117/12.208476 0277-786X Google Scholar
T. French,
B. Bailey,
D. P. Stumbo, and
D. Modlin,
“A time-resolved fluorometer for high-throughput screening,”
3603 272
–280
(1999) Google Scholar
D. S. Elson,
J. Siegel,
S. E. Webb,
S. Lévêque-Fort,
M. J. Lever,
P. M. French,
M. Lauritsen,
M. Wahl, and
R. Erdmann,
“Fluorescence lifetime system for microscopy and multiwell plate imaging with a blue picosecond diode laser,”
Opt. Lett., 27
(16), 1409
–1411
(2002). 0146-9592 Google Scholar
R. Kohlrausch, Ann. Phys., Lpz., 12 393
(1847). Google Scholar
P. E. Fraser,
D. S. Yang,
G. Yu,
L. Levesque,
M. Nishimura,
S. Arawaka,
L. C. Serpell,
E. Rogaeva, and
P. St. George-Hyslop,
“Presenilin structure, function and role in Alzheimer disease,”
Biochim. Biophys. Acta, 1502
(1), 1
–15
(2000). 0006-3002 Google Scholar
D. J. Selkoe,
“Presenilin, notch, and the genesis and treatment of Alzheimer’s disease,”
Proc. Natl. Acad. Sci. U.S.A., 98
(20), 11039
–11041
(2001). https://doi.org/10.1073/pnas.211352598 0027-8424 Google Scholar
M. E. Fortini,
“Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling,”
Nat. Rev. Mol. Cell Biol., 3
(9), 673
–684
(2002). 1471-0072 Google Scholar
D. J. Selkoe,
“Amyloid beta-protein and the genetics of Alzheimer’s disease,”
J. Biol. Chem., 271
(31), 18295
–18298
(1996). 0021-9258 Google Scholar
A. Lleo,
O. Berezovska,
J. H. Growdon, and
B. T. Hyman,
“Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with ps-1 mutations,”
Am. J. Geriatr. Psychiatry, 12
(2), 146
–156
(2004). 1064-7481 Google Scholar
A. Lleo,
O. Berezovska,
L. Herl,
S. Raju,
A. Deng,
J. Bacskai,
M. P. Frosch,
M. Irizarry, and
B. T. Hyman,
“Nonsteroidal anti-inflammatory drugs lower beta42 and change presenilin 1 conformation,”
Nat. Med., 10
(10), 1065
–1066
(2004). 1078-8956 Google Scholar
O. Berezovska,
A. Lleo,
L. D. Herl,
M. P. Frosch,
E. A. Stern,
B. J. Bacskai, and
B. T. Hyman,
“Familial Alzheimer’s disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein,”
J. Neurosci., 25
(11), 3009
–3017
(2005). https://doi.org/10.1523/JNEUROSCI.0364-05.2005 0270-6474 Google Scholar
N. PanchukVoloshina,
R. P. Haugland,
J. Bishop Stewart,
M. K. Bhalgat,
P. J. Millard,
F. Mao,
W.-Y. Leung, and
R. P. Haugland,
“Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates,”
J. Histochem. Cytochem., 47
(9), 1179
–1188
(1999). 0022-1554 Google Scholar
O. Berezovska,
B. J. Bacskai, and
B. T. Hyman,
“Monitoring proteins in intact cells,”
Sci. Aging Knowledge Environ., 2003
(23), PE14
(2003). Google Scholar
D. J. Selkoe,
M. B. Podlisny,
C. L. Joachim,
E. A. Vickers,
G. Lee,
L. C. Fritz, and
T. Oltersdorf,
“Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kiloDalton membrane-associated proteins in neural and nonneural tissues,”
Proc. Natl. Acad. Sci. U.S.A., 85
(19), 7341
–7345
(1988). https://doi.org/10.1073/pnas.85.19.7341 0027-8424 Google Scholar
T. Förster,
“Zwischenmolekulare enerergiewanderung und fluoreszenz,”
Ann. Phys. Lpz., 2 55
–75
(1948). Google Scholar
G. Nishimura and
M. Tamura,
“Artefacts in the analysis of temporal response functions measured by photon counting,”
Phys. Med. Biol., 50
(6), 1327
–1342
(2005). https://doi.org/10.1088/0031-9155/50/6/019 0031-9155 Google Scholar
B. van der Meer,
G. Coker Ill, and
S.-Y. Chen, Resonance Energy Transfer: Theory and Data, VCH Publishers, New York (1994). Google Scholar
W. Press,
B. Flannery,
S. Teukolsky, and
W. Vetterling, Numerical Recipes in C: The Art of Scientific Computing, Cambridge University Press, NewYork (1992). Google Scholar
F. Alvarez,
A. Alegra, and
J. Colmenero,
“Relationship between the time-domain Kohlrausch–Williams-watts and frequency-domain Havriliak–Negami relaxation functions,”
Phys. Rev. B, 44
(1), 7306
–7312
(1991). https://doi.org/10.1103/PhysRevB.44.7306 0163-1829 Google Scholar
J. C. Phillips,
“Stretched exponential relaxation in molecular and electronic glasses,”
Rep. Prog. Phys., 59
(9), 1133
(1996). https://doi.org/10.1088/0034-4885/59/9/003 0034-4885 Google Scholar
R. Chen,
“Apparent stretched-exponential luminescence decay in crystalline solids,”
J. Lumin., 102–103 510
–518
(2003). 0022-2313 Google Scholar
K. C. B. Lee,
J. Siegel,
S. E. D. Webb,
S. Lévêque-Fort,
M. Cole,
R. Jones,
K. Dowling,
M. J. Lever, and
P. M. W. French,
“Application of the stretched exponential function to fluorescence lifetime imaging,”
Biophys. J., 81
(3), 1265
–1274
(2001). 0006-3495 Google Scholar
|



